Reports: ND456668-ND4: Superstrained C5 Hydrocarbons
 printer friendly
printer friendly1. Chlorination of BCP. 2-Chloro and 2,2-dichloro BCP were already known.[1] We now prepared and characterized the new 2,2,4-trichloro and 2,2,4,4-tetrachloro derivatives.
The starting material, bicyclo[1.1.1]pentane-1,3-dicarboxylic acid (1) was prepared from 2 according to published procedures (Scheme 1).[2],[3]
Scheme 1. Synthesis of dicarboxylic acid 1. |
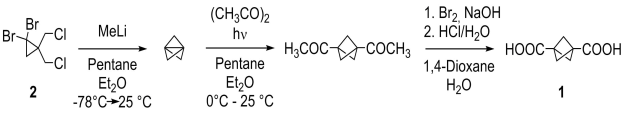
The diacid 1 was converted to its dichloride with SOCl2 and chlorinated with Cl2 in CCl4 under irradiation with a 500 W halogen lamp. We were unable to find reaction conditions yielding only 4 and the trichloro derivative 5 was always present in the reaction mixture (Scheme 2, route A).
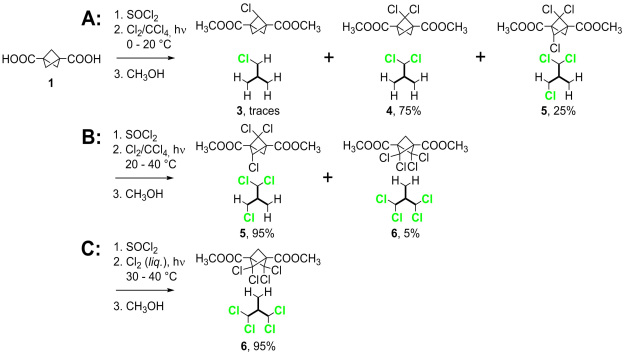
Scheme 2. Three synthetic routes leading to 4, 5, and 6. |
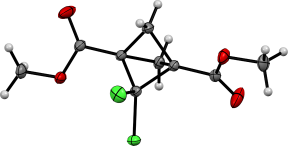
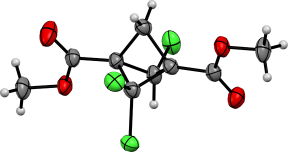
The products were separated using column chromatography and characterized by X-ray diffraction (Figure 1).
Figure 1. ORTEP representations of 4 and 5.
Slightly elevated temperature (20 - 40 °C) and extended reaction times (ca 4 days) lead to nearly quantitative formation of 5, with traces of 6 (Scheme 2, route B).
Targeted synthesis of 6 required much harsher reaction conditions: liquid chlorine, 40 °C, irradiation with 500 W halogen lamp and reaction time reaching 40 hours. Even under these conditions, no rearrangement of BCP cage was observed and 6 was formed as the only product in nearly quantitative yield (Scheme 2, route C). This clear colorless liquid slowly solidified, but attempts to obtain a single crystal have not yet succeeded. The structure was confirmed by 13C NMR.
All attempts to introduce five or six chlorine atoms into the BCP cage have failed so far.
2. Protection of Carboxylate Groups in Positions 1 and 3. Not surprisingly, preliminary experiments showed that n-butyllithium reacts not only with the CCl2 bridge in the lithium salt of the diacid derived from 4 but also with the carboxylate groups themselves, and that it will be necesary to protect the latter. The methyl and the t-butyl esters of the diacid reacted with n-butyllithium as well. In all these reactions, the CCl2 group showed encouraging signs of conversion to a carbenoid followed by insertion into the neighboring CH2 group, and it was clear that a better protecting group needs to be found. An attempt to convert the diacid to a bis(trifluoromethyl) derivative by reaction with SF4 failed.
Our next choice were cyclic orthoesters. To test their suitability, a cyclic orthoester of 1,3-adamantanedicarboxylic acid 7 was chosen as the model compound. Following a literature procedure, It was obtained[4] by treatment of the double acyl chloride with 3-methyl-3-oxetanemethanol to give the bisoxetane derivative 8. Rearrangement to the cyclic orthoester 9 was achieved using BF3.Et2O in dichloromethane in quantitative yield (Scheme 3).[5]
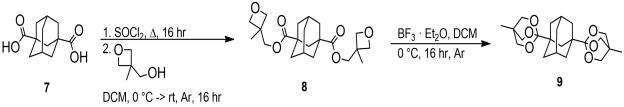
Scheme 3. Preparation of cyclic orthoester 9. |
The orthoester 9 resisted excess n-BuLi in THF even upon prolonged stirring at room temperature, suggesting that this type of structure was a good choice as a protective group. Then, a cyclic orthoester 10 of the 2,2-dichlorobicyclo[1.1.1]pentane-1,3-dicarboxylic acid derived from the diester 4 was synthesized in a similar manner (Scheme 4).
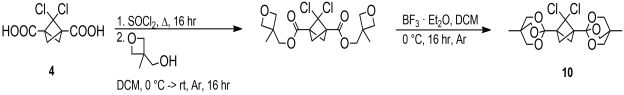
Scheme 4. Synthesis of cyclic orthoester 10. |
Unfortunately, the rearragement reaction was not clean and the byproducts hindered purification. The same problem was encountered with other Lewis acids (AlMe3, TMSOTf, and CF3SO3H), and literature analogies were found.[6],[7],[8] To avoid these difficulties, we turned to oxazolines as an alternative protection group. First, an oxazoline based on 1-adamantane carboxylic acid 11 was synthesized as a model by treating its acyl chloride with excess 2-amino-2-methyl-1-propanol dichloromethane and then with aqueoius NaOH to yield the oxazoline 12.[9] Alternatively, 12 was prepared by refluxing the acid chloride with excess 2-amino-2-methyl-1-propanol in tetrahydrofuran, followed by treatment with aqueous sodium hydroxide. The yield was quantitative in both cases (Scheme 5).
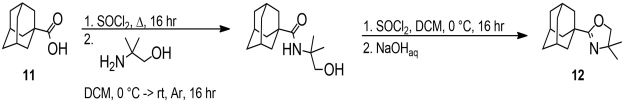
Scheme 5. Preparation of the oxazoline 12. |
The oxazoline 12 resists excess n-BuLi in THF after prolonged stirring at ambient conditions. Then, bisoxazoline 13 derived from the parent diacid 1 was prepared in a similar manner (Scheme 6).
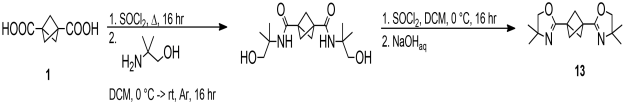
Scheme 6. Preparation of bisoxazoline 13 based on the bicyclo[1.1.1]pentane-1,3-dicarboxylic acid 1. |
The bisoxazoline 13 resisted excess n-BuLi in THF almost entirely at temperatures below -40̊ C, suggesting that the oxazoline moiety is a good candidate for a protective group for chlorinated BCP derivatives. Preparation of bisoxazolines 14 and 15 derived from 4 and 5, respectively, was attempted in a similar manner and hydroxyamides were obtained in quantitative yields. However, their conversion to chlorides took much longer than for the parent BCP derivative. Moreover, the bisoxazolines 14 and 15 appeared to be more prone to hydrolysis during workup. These differences can be attributed to the increased acidity of the highly chlorinated diacids. The oxazolines 14 and 15 were therefore prepared using an alternative method in which the amide is dissolved in a mixture of MeCN, NEt3 and CCl4 and treated with excess PPh3[10] (Scheme 7).
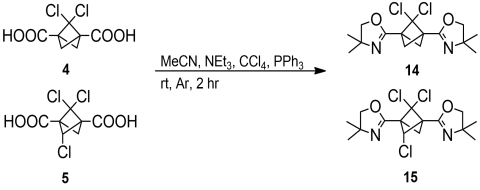 |
Scheme 7. Synthesis of bisoxazolines 14 and 15. |
At this point, we are ready to treat the chlorinated double oxazolines with n-butyllithium at low temperature in the hope of producing carbenoids, the products of whose thermal decomposition to carbenes will then be examined.
LITERATURE
[1]. Robinson, R. E.; Michl, J. J. Org. Chem. 1989, 54, 2051-2053.
[2]. Kaszynski, P.; Michl, J. J. Org. Chem. 1988, 53, 4594-4596.
[3]. Kaleta, J.; Mazal, C. Org. Lett. 2006, 8, 749-752.
[4]. Corey, E.J.; Raju, N. Tetrahedron Lett. 1983, 24(50), 5571-5574.
5. Meyers, A. I.; Temple, D. L.; Haidukewych, D.; Mihelich, E. D. J. Org. Chem. 1974, 39, 2787-2793.
[6]. Kanoh, S.; Naka, M.; Nishimura, T.; Motoi, M. Tetrahedron 2002, 58, 7049-7064.
[7]. Kanoh, S.; Naka, M.; Yokozuka, T.; Itoh, S.; Nishimura, T.; Honda, M.; Motoi, M.; Matsuura, N. Macromol. Chem. Phys. 2002, 203, 511-521.
[8]. Kanoh, S.; Nishimura, T.; Tsuchida, T.; Senda, H.; Motoi, M.; Takani, M.; Matsuura, N. Macromol. Chem. Phys. 2001, 202, 2489-2503.
[9]. Meyers, A. I.; Temple, D. L.; Haidukewych, D.; Mihelich, E. D. J. Org. Chem. 1974, 39(18), 2787-2793.
[10]. Kawasaki, K.; Katsuki, T. Tetrahedron 1997, 53(18), 6337-6350.










