Reports: DNI655181-DNI6: Chemically Targeted X-ray Spectroscopic Studies of Chromium-Catalyzed Ethylene Trimerization Reactions
 printer friendly
printer friendlyDuring the 2016–2017 funding period we pursued studies of homogeneously catalyzed reactions mediated by the earth abundant transition metals copper and iron. This marked a departure from the goal proposed at the outset of this project, which was to investigate active species within ethylene trimerization reactions employing chromium diphosphene precatalysts. Before the grant was activated, we had conducted a rigorous calibration study1 of chromium V2C XES. This study found that although the technique is highly sensitive to the identities of coordinated ligands, the other ligands within a complex will perturb V2C XES band energies such that substantial overlap precludes definitive assignment of bands to individual ligands (Figure 1).
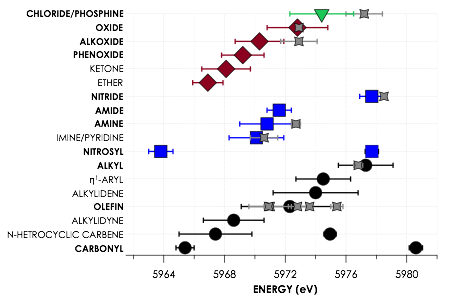
Figure 1. Plotted spectrochemical series displaying the average energies of experimentally calibrated, DFT-calculated Kβ″ V2C XES features for ligands grouped by donor atom. Error bars represent doubled standard deviations. Gray points indicate experimental Kβ″ energies corresponding to the indicated ligands, with error bars derived from fits to experimental spectra. Ligands indicated in bold are predicted to significantly contribute intensity to Kβ″ regions, while nonbolded ligands are expected to be difficult to resolve with current experimental limitations. Adapted from Inorg. Chem. 2014, 54, 205–214.
In order to achieve progress toward our goal of developing X-ray spectroscopies for use in characterizing species relevant to homogeneous catalysis and small molecule activation, we elected to shift focus to reactions with narrower parameter spaces. Two aims were pursued: a spectroscopy-based explanation for the reactivity of Cu/Aminoxyl (R2NO·) complexes in selective alcohol oxidation, and the role of redox-active imide/amide ligands in C–H amination reactions.
Cu/Aminoxyl complexes were furnished by the laboratory of our collaborator, Prof. Shannon Stahl (UW-Madison). Using Cu L2,3-, Cu K-, and Cl K-edge X-ray absorption spectroscopy (XAS), my laboratory showed that coordination of an aminoxyl ligand to Cu(II) causes the coordinated aminoxyl (TEMPO or ABNO) to exhibit oxoammonium (R2N=O+) character (Figure 2). This is noteworthy because oxoammonium ions are well-known to effect alcohol oxidation, while neither Cu(II) complexes nor free aminoxyl mediate these transformations. This oxoammonium character, exposed through metal-ligand cooperation, is thus implicated in the ability to effect 2-electron oxidation of alcohols to aldehydes or ketones. This work was recently published in J. Am. Chem. Soc. 2017, 138, 13507–13517.
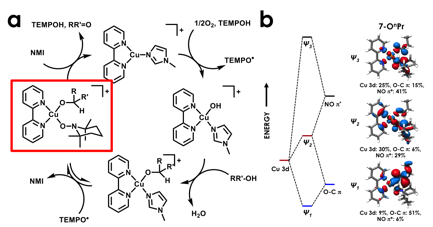
Figure 2. (a) Minimal mechanism for alcohol oxidation by Cu/TEMPO/2,2¢-bipyridine (bpy) catalyst system. Adapted from J. Am. Chem. Soc. 2014, 136, 12166−12173. (b) Experimentally-validated frontier molecular orbital diagram for CuII/TEMPO/bpy/alkoxide intermediate implicated in alcohol oxidation, showing that the lowest unoccupied molecular orbital's hole density is ligand-localized, indicating a high degree of CuI–oxoammonium character.
In collaboration with the laboratory of Theodore Betley (Harvard), we carried out XAS studies of dipyrrin-supported Fe–imide and –iminyl complexes. Using N K-edge XAS, we directly demonstrated the presence of a sub-valent iminyl ligand in the inner sphere of Fe, and were able to quantify different degrees of N hole character in species with different R-groups attached to the iminyl (Figure 3). The electronic structure picture was then used to demonstrate that Fe–iminyl species are far more effective mediators of C–H amination than the corresponding Fe–imides, whose N–R fragments are closed-shell. This work has been accepted for publication in J. Am. Chem. Soc.
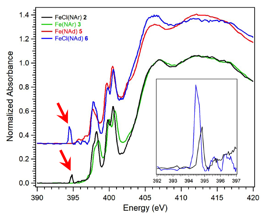
Figure 3. N K-edge XAS of Fe–imide (Fe(NR)) and Fe–iminyl (FeCl(NR)) complexes used in C–H amination reactions. The red arrows indicate transitions unique to the iminyl complexes that are assigned as N 1s ¨ iminyl N 2p. Adapted from J. Am. Chem. Soc. 2017, DOI: 10.1021/jacs.7b08714.
In addition to the primary research articles described above, this ACS PRF DNI grant was credited in part for the ability to assemble a perspective article for ACS Catalysis meant as an introduction to information content potentially available to researchers in homogeneous catalysis should they pursue synchrotron-based X-ray spectroscopic studies. The corresponding publication, ACS Catalysis 2017, 7, 1776–1791, has been widely disseminated through social media, and was featured in the August 2017 virtual issue of Chem. Mater.
The majority of expenditures in the 2016–2017 funding period went to support a graduate student, Mr. James Lukens. However, because of high costs associated with support of Ph.D. students at Cornell, Mr. Lukens' support was offset by one semester of a TA-ship. Regardless, he has been able to develop considerable expertise as an X-ray spectroscopist. Moreover, Mr. Lukens has developed proficiency in chemical synthesis and electronic structure calculations. During this period, supplementary funds from my NSF CAREER grant allowed Mr. Lukens to travel to the Stanford Synchrotron Radiation Lightsource to carry out experiments. In addition, Mr. Lukens traveled to the ACS Fall 2017 National Meeting in Philadelphia, where he presented a poster on his ACS PRF-supported work.










