Reports: DNI557079-DNI5: Effects of Zeolite Framework Topology for Conversion of Methanol/Ethanol to Hydrocarbons: Towards Computer-Directed Synthesis
 printer friendly
printer friendlyProton-form zeolites can be used to catalytically upgrade methanol into gasoline-range hydrocarbons or aromatics in methanol-to-hydrocarbons (MTH) processes. The pore structure surrounding the acid of proton-form zeolite catalysts governs reaction rates and selectivities through stabilization (or destabilization) of kinetically relevant transition states and critical intermediates. Experimental kinetic studies of MTH processes are convoluted by the complex reaction networks and instability of rates with time-on-stream. Theoretical approaches such as density functional theory (DFT) can be employed in combination with experiments to model these complex reaction networks, determine how zeolite topology governs these processes, and ultimately identify novel catalysts for improved MTH performance.
MTH turnover rates increase and subsequently decrease with time-on-stream (showing auto-catalytic behavior and deactivation). A pool of hydrocarbon species is formed that act as cocatalysts by rapidly converting methanol through facile C–C bond formation in the olefin and aromatic cycles (Fig. 1), resulting in auto-catalysis.
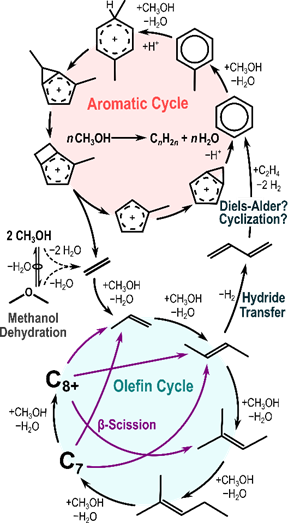
Figure 1. Dual cycle mechanism of alkene formation during MTO.
Methanol forms surface methoxy species which methylate olefins repeatedly until they reach a size capable of cracking into smaller products in the olefin cycle. Methanol and hydrocarbon species can also undergo hydride transfer reactions, forming alkanes as well as formaldehyde and dienes, the latter of which can lead to the formation of aromatic species. These aromatics can form poly-aromatics which deactivate catalysts, and can also cocatalyze methanol conversion into light alkenes though several possible mechanisms (Figure 2). The side-chain mechanism involves direct methylation of an exocyclic C-substituent until the side-chain is large enough to detach as an olefin. The expansion-contraction and contraction-expansion mechanisms form side chains through methylation and manipulation of the aromatic ring, and these cycles will therefore incorporate ring C-atoms into product alkenes.
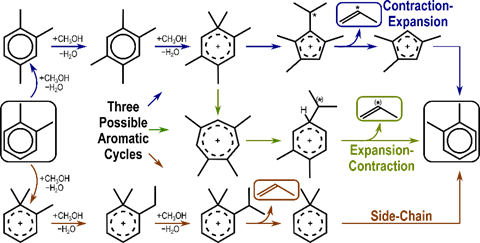
Figure 2. Mechanisms for alkene formation through contraction-expansion, expansion-contraction, and side-chain routes. Each cycle can form C2–C4 alkenes using many possible aromatic species (propene formation from o-xylene shown). An (*) represents incorporation of a ring C-atom.
Each of the three mechanisms described in Fig. 2, however, can occur via multiple pathways, and the preferred pathway and its kinetically relevant step depends on the alkene being formed and the aromatic co-catalyst. Ethylene formation, for example, prefers to proceed via a side-chain mechanism 1,2,4-trimethylbenzene is the co-catalyst (Fig. 3a). When using benzene as the co-catalyst, however, ethylene is formed via the ring expansion-contraction pathway (Fig. 3b) because the nature of the co-catalyst has a significant impact on the activation barriers and reaction energies of each step within these cycles. Unfortunately, the effects of methylation depend on the exact reaction being studied (e.g., arene methylation, ring contraction) and do not exhibit simple linear trends which could be easily regressed to estimate energetics (Fig. 4)—instead, we must examine all co-catalysts for all elementary steps involved in MTH processes.
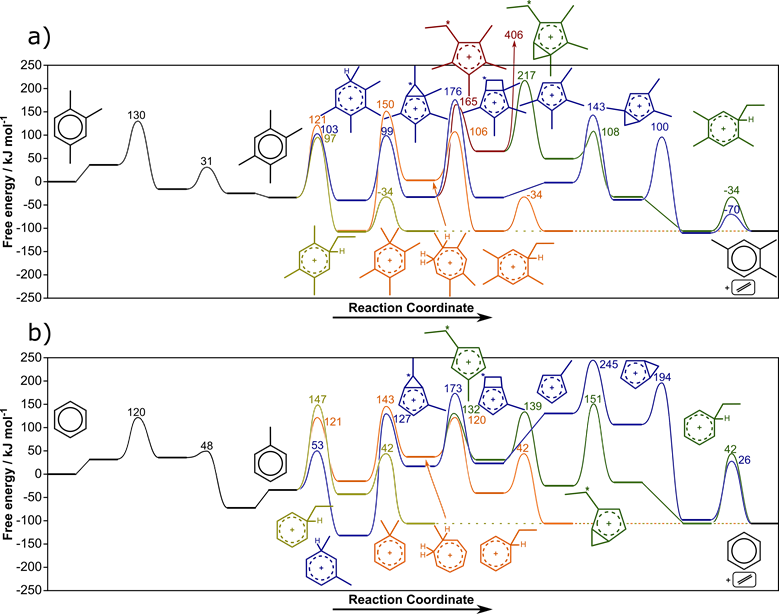
Figure 3. Ethylene formation from a) 1,2,4-trimethylbenzene and b) benzene via side-chain (chartreuse), ring expansion-contraction (orange), contraction-dealkylation-expansion (red), contraction-expansion-dealkylation (green), and contraction followed by the formation of a bicyclic intermediate (blue).
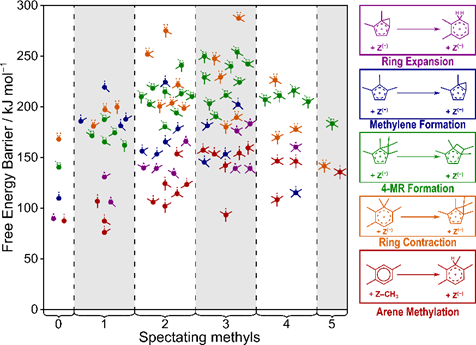
Figure 4. Effects of methylation of several elementary steps involved in aromatic cycles of MTH reactions.
The substantial number of possible reaction pathways present in the aromatic cycle precludes a complete ‘graphical’ analysis of the type shown in Figs. 3 and 4, as often done in DFT studies of reaction pathways. To form propene from 1,2,4-trimethylbenzene, for example, there are 42 different pathways; to form propene from any co-catalyst, there are ~485 pathways. Thus, a kinetic Monte Carlo (KMC) program is being developed as part of this proposal to stochastically determine the formation rates of C2-C4 alkanes, and the most productive pathways and co-catalysts involved at a range of temperatures and reagent pressures.
All data shown in Figs. 3 and 4 are for reactions carried out at the T-11 site of MFI (out of 12 crystallographically unique T-sites). Changing the local environment around reaction intermediates and critical transition states can have dramatic effects on reaction energetics and pathway selectivity. The transition state for zeolite methylation from CH3OH, for example, has energies that vary by ~150 kJ mol−1 in MFI when the orientation or acid site location is changed (Fig. 5), with other frameworks (CHA, BEA) exhibiting similar trends. The most stable transition state locations within these frameworks involve H-bonding to the cage and optimal van der Waals stabilization, while less stable structures reside either in large environments that are incapable of solvating the transition states, or in small environments that must restructure at significant cost to accommodate the transition state.
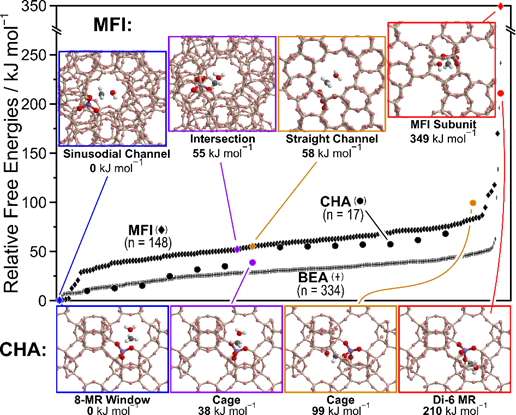
Figure 5. Effects of transition state orientation and location for CHA, MFI, and BEA zeolite frameworks. Specific structures for the MFI and CHA shown in insets.
Evaluating every critical transition state at every site with multiple configurations using DFT is not computationally tractable. Therefore, we are developing state specific force fields (SSFF) capable of modeling adsorbate-framework interactions (i.e., dispersion, Pauli repulsion, electrostatic interactions, and H-bonding, Fig. 7) for key intermediates and transition states. Once generated, these SSFF will be used to predict optimal structures that will then be calculated using DFT. To identify potential catalysts with improved selectivity towards light olefins and lower rates of deactivation, MTH will first be evaluated at a single site in MFI using DFT to determine critical intermediates and transitions states. The large and complex reaction networks will then be analyzed using KMC simulations, and, once key structures have been determined, every site in MFI will be evaluated. SSFF will be created for every adsorbate using DFT results, and will be applied to other synthesized (>200) and predicted (>300,000) frameworks.
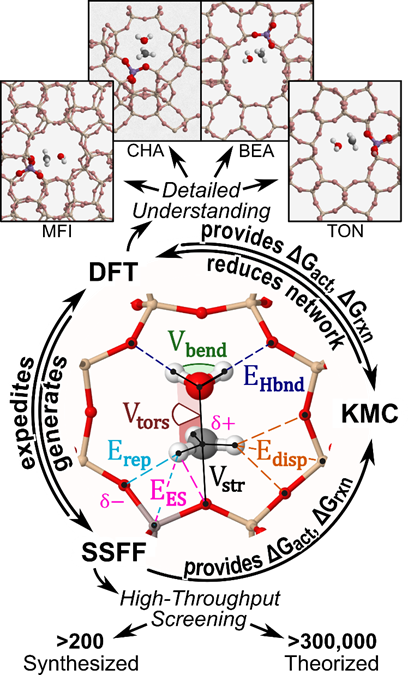
Figure 6. Multiscale modeling approach for screening zeolites using a combination of DFT, KMC, and SSFF.
This proposal funded two Ph.D. candidates last year (each at 50% appointment) and will continue to fund them next year. It also funded one students’ travel to the North American Meeting of NACS in Denver where he presented a poster. This proposal, furthermore, has helped develop ideas and provided preliminary data for my NSF CAREER proposal, submitted this past summer, and still under review.










