Reports: ND356871-ND3: Activation of Carbon Dioxide with Antimony Lewis Acids
 printer friendly
printer friendlyIn the first year of this award, we have been able to address some of the central goals of our research project by investigating the synthesis of new antimony-based Lewis acids, with the view of employing such Lewis acids for the activation of small molecules including CO2.
With this objective in mind, we have targeted stibonium cations whose Lewis acidity is enhanced by the presence of a halogen substituent directly bound to antimony. Starting from Ph3Sb(OTf)2 and Mes3Sb(OTf)2, we successfully prepared the triflate derivatives Ph3SbF(OTf) and Mes3SbF(OTf). We also synthesized the hexachloroantimonate salt of [Mes3SbCl]+ (2), an analog of the known [Ph3SbCl][SbCl6] (1) (Figure 1A).1 We obtained single crystals of these compounds which allowed us to examine the structure of these salts in the solid state. While a direct interaction is observed between the anion and the stibonium center in most compounds investigated including 1, compound 2 exists as an ionic solid with the four coordinate [Mes3SbCl]+ stibonium cation separated from the [SbCl6]- anion (Figure 1B). The structural difference observed between the two hexachloroantimonate derivatives 1 and 2 is ascribed to the increased steric protection provided by the larger mesityl substituents. To understand how these structural differences affect the properties of these antimony species, we have compared their catalytic activity in two simple reactions, namely the polymerization of THF and the Friedel-Craft dimerization of 1,1-diphenlyethylene (Figure 1C). These studies show that 1 is the most active catalyst for both reactions thus suggesting that the reactivity of these species is controlled both by the coordinating nature of the counteranions and the steric accessibility of the reactive antimony center. Finally, we have correlated the high Lewis acidity of these stibonium cations to the presence of a low lying molecular orbital of s*(Sb-Cl) character centered in the antimony atom. We are currently working to apply these new Lewis acids to the activation of CO2.
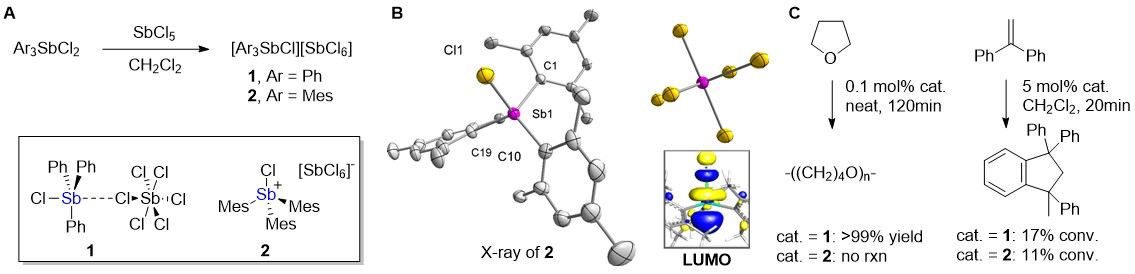
Figure 1. A: Synthesis of 1 and 2. B: Structure of 2 in the crystal with the LUMO of 6 in the inset. C: Catalytic properties of 1 and 2.
We have also probed whether cooperative effects could be used to enhance the Lewis acidity of antimony derivatives. These efforts have led us to target a bidentate Lewis acid in which the two antimony centers are connected by a 1,8-dibromotriptycenediyl backbone. Starting from 1,8-dibromotriptycene, distibine 3 was first obtained and subsequently treated with o-chloranil to afford the distiborane 4 (Figure 2A), the structure of which was confirmed by X-ray diffraction.2 We decided to probe the Lewis acidity of this compound using the fluoride anion as a probe. We found that it cleanly reacts with [NnBu4][Ph3SiF2] (1 equiv) in CH2Cl2 to afford the fluoride complex ([NnBu4 ][4-m2-F]) whose crystal structure confirmed the presence of a Sb-F-Sb anion chelate motif (Figure 2B). Moreover, the 1H NMR spectrum of [4-m2-F]-, indicated that the proton bound to the upper triptycene bridgehead carbon atom is coupled to the fluorine nucleus (1JHF = 4.9 Hz) suggesting the existence of a C-H···F hydrogen bond. This view is supported by the crystal structure of the complex which shows a short C19-F1 distance of 2.915(4) Å. This C-H···F hydrogen bond interaction has also been studied using the Natural Bond Orbital method. Application of this method shows a weak donor-acceptor lp(F)"s*(bridgehead C-H) interaction of E(2) = 5.9 kJ/mol, a value that falls in the expected range for a weak hydrogen bond. These results led us to conclude that the 1,8-triptycenediyl backbone is especially well suited for anion chelation. This view was reinforced through a comparison with a related system in which the two stiborane moieties are connected by a 1,8-dimethylxanthenediyl backbone.

Figure 2. A: Synthesis of 4 and [NnBu4][4-m2-F]. B: Solid state structure of [2-m2-F]-. The inset shows the NBO plot of the lp(F)"s*(bridgehead C-H) donor-acceptor interaction.
Finally, funds provided by the PRF have allowed us to explore intramolecular frustrated Lewis pairs based on Lewis acidic moieties other than antimony. In particular, we have investigated the synthesis of ortho-phenylene-based bifunctional compounds featuring a tritylium cation as a Lewis acid and a phosphine as a Lewis base.3 We observed that these compounds form four-membered phosphonium species (such as 5+ and 6+) as a result of an intramolecular Lewis acid-Lewis base neutralization. The four membered phosphonium cations showed no reactivity toward small molecules such as CO2 indicating that the stability of the P"C+ bond is inhibitory.
References
1. Yang, M.; Gabbaï, F. P. "Synthesis and Properties of Triarylhalostibonium Cations" Inorg. Chem. 2017, 56, 8644-8650.
2. Chen, C.-H.; Gabbaï, F. P. "Fluoride Anion Complexation by a Triptycene-Based Distiborane: Taking Advantage of a Weak but Observable C−H⋅⋅⋅F Interaction" Angew. Chem. Int. Ed. 2017, 56, 1799-1804.
3. Chansaenpak, K.; Yang, M.; Gabbaï, F. P. "Attempted synthesis of ortho-phenylene phosphino-tritylium cations" Phil. Trans. R. Soc. A 2017, 375, 20170007










