Reports: UR156692-UR1: Exploring the Potential of an Acid-Initiated Vinylogous Aldol Reaction to Form All-Carbon Quaternary Stereocenters
 printer friendly
printer friendlyThe stereoselective synthesis of all carbon quaternary stereocenters (ACQSs) is an important challenge in modern organic chemistry. Our interest in exploring the assembly of ACQSs derives from an unexpected intramolecular vinylogous aldol (IVAR) reaction that took place when attempting to hydrolyze the methyl dienol ether resident in 1 (with n = 3 and R = Me) under catalytic acid conditions in wet THF to give 2E (n = 3) and 2Z (n = 3) in a 10 : 1 diastereomeric ratio, respectively (Scheme 1).
We began our exploration of this IVAR by addressing the question of what size rings can be formed using this approach (Scheme 1). To that end, we prepared a number of substrates (1) bearing alkyl or aryl dienol ethers linked to an aldehyde by homologous methylene tethers.
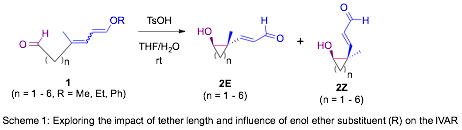
The IVAR substrates (1) are prepared from readily available straight-chain diols (3, Scheme 2).
Our established protocol to convert the intermediate methyl ketones 4 into the corresponding alkyl/aryl dienol ethers makes use of phosphonates 5 in a Horner-Emmons Wittig reaction (Compt. Rend. 1965, 261 (14), 2679-81). While this approach allows direct introduction of the desired functionality, the approach lacks stereoselectivity and generality. Of particular concern, in our hands, the preparation of 5a has proven elusive and limits access to methyl dienol ethers to lengthier approaches (Scheme 3). In contrast, 5b ad 5b are known to the literature, are readily prepared in high yield, and provide modest yields of ethyl and phenyl dienol ethers when employed for intermediates with n = 2 – 5. The reaction has yet to function with the shortest (n = 1) and longest (n = 6) substrates, again highlighting its shortcomings.
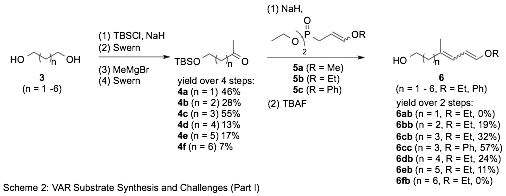
To address these limitations we are investigating a number of longer approaches that may have improved efficiency, be more general, and allow for the isolation of single isomers of dienol ethers 6 (Scheme 3). For example, we have made progress exploring two potential alternatives to form dienol ethers (8) from the model substrate 2-heptanone (7).

We have pushed forward with primary alcohols (6) with tether lengths from n = 2 to n = 5 which were oxidized to the requisite IVAR substrates (1) and exposed toluenesulfonic acid in aqueous THF to determine their propensity to cyclize (Scheme 4). Thus far we have observed the operation of the IVAR to form 4-, 5- and 6- membered carbocyclic aldols with ACQSs.
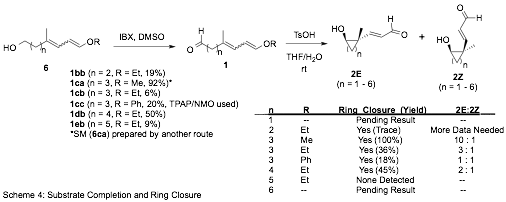
Only a trace of the cyclobutane products (2E/2Z, n = 2) was observed and additional material will be prepared to determine the diastereoselectivity of the process. Formation of cycloheptane aldol products (n = 5) was not observed. The cyclopentane derivatives (n = 3) from 1ca, 1cb, and 1cc revealed the importance of the structure dienol ether substituent (R). When R = Me a 10 : 1 ratio of 2E : 2Z, respectively, was realized. With R = Et the diastereoselecivity dropped to 3 : 1 for 2E : 2Z, respectively, and diastereoselectivity was lost altogether when R = Ph. Given its relative remoteness to the reacting centers, it was a surprise to us that the diastereoselectivity was so dramatically influenced by the nature of R. However, given its evident role, we are compelled to find a synthesis that allows access to VAR substrates 1 with a variety of R groups and with a single stereochemical disposition of the resident alkenes.
One additional VAR substrate (9), with a three-carbon tether bearing a methyl group on the central carbon (C4), was prepared by a route analogous to that used for substrates 1 (Scheme 5).
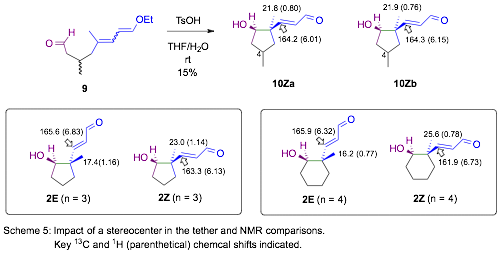
The presence of the stereocenter in the tether has a profound impact on the diastereoselectivity of the process and yielded only Z diastereomers (with the C1-hydroxyl and the C2-acryladehyde on the same face). C4 stereoisomers were observed in a 2 : 1 ratio although more work is needed to sort out which of the two stereoisomers is dominant (10Za or 10Zb). NMR analysis provides strong support of the stereochemistry around the newly formed bond, via g-substituent effects on 13C chemical shifts, a lack of informative NOE correlations to the C4 methyl group has prompted us to consider other approaches to determine its stereochemistry.
The research results described in this report were generated by nine different students at different stages of their undergraduate chemistry education. Of the nine, four have now graduated and are in excellent chemistry graduate programs (2 students), working as a post-baccalaureate researcher in a graduate lab (1 student), or are in the process of applying to graduate school (1 student). In addition to making contributions to this project, these senior students helped mentor the 5 current research students. We are grateful for the support of the ACS Petroleum Research Fund and hope it is evident that this grant has had a strong and positive impact on the students and PI involved in this project.










