Reports: ND156369-ND1: Hydrogenation of C(sp3)-O bonds Catalyzed by Acidic Group 9 Hydride Complexes
 printer friendly
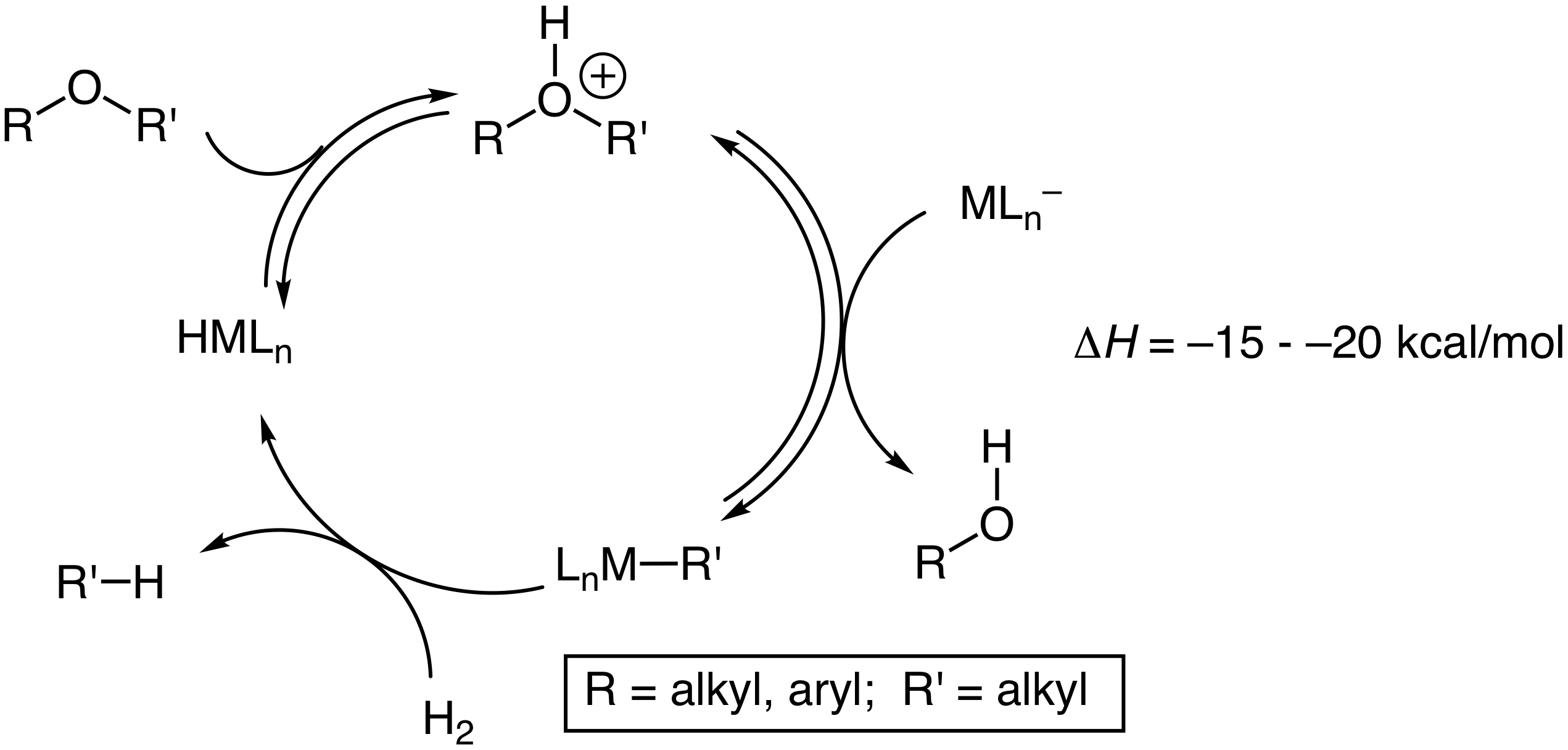
printer friendlyIntroduction. The overarching objective of this project is the synthesis of group 9 hydride complexes bearing p-acceptor phosphine ligands to be employed as catalysts for the hydrogenolysis and silyative hydrogenolysis of C(sp3)–O bond of dialkyl and alkyl aryl ethers. The targeted and mechanistically distinct catalytic cycle involves 1) proton transfer from the acidic catalyst to the ether oxygen atom, 2) nucleophilic displacement of the protonated carbinol with the resulting transition metal anion, and 3) hydrogenolysis of the resulting metal alkyl complex with H2 to regenerate the metal hydride catalyst (Scheme 1).
Scheme 1
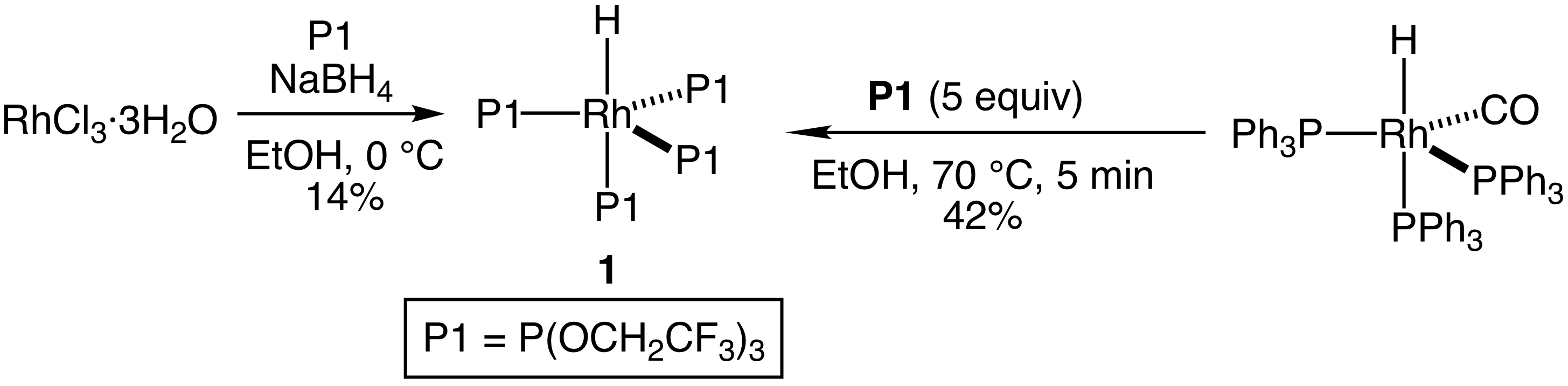
Progress toward objectives. Our efforts in the current funding period focused initially on evaluating the reactivity of the rhodium tetrakis(flourinated phosphite) hydride complex HRh[P(OCH2CF3)3]4 (1), which we were able to isolate in 14% yield from reaction of rhodium trichloride hydrate, sodium borohydride and P(OCH2CF3)3 (Scheme 2). Because our focus was to be on stoichiometric reactions of 1, given the expense of rhodium, we sought to develop a more efficient preparation for 1. To this end, we have found that reaction of HRh(CO)[(PPh3)3] with P1 in ethanol at 70 °C for 5 min led to the isolation of 1 in 42% yield as analytically pure material (Scheme 1).
Scheme 2
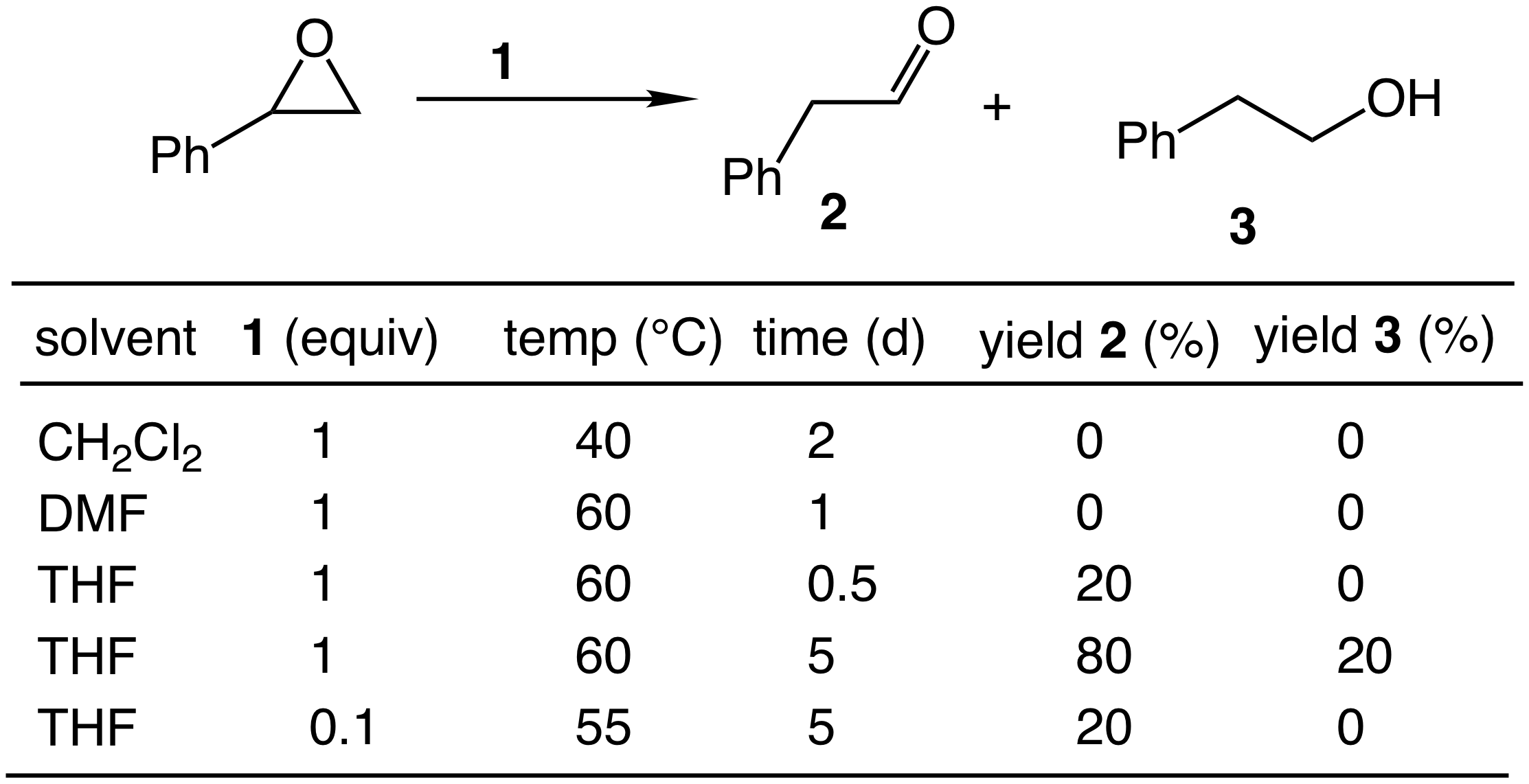
With an efficient synthesis of 1 in hand, we turned our attention to an evaluation of the reactivity of 1 toward C–O bond cleavage and, to this end, we investigated the reaction of styrene oxide with 1 under stoichiometric conditions. No consumption of styrene oxide was observed upon reaction with 1 in either methylene chloride at 40 °C or DMF at 60 °C for 1-2 days (Table 1). However, THF proved a more suitable solvent and reaction of styrene oxide with 1 in THF at 60 °C for 5 days led to complete conversion to form 2-phenylacetaldehyde in 80 % yield along with 2-phenylpropanol in 20% yield (Table 1). These outcomes were in accord with a mechanism involving initial proton transfer from 1 to styrene oxide followed by nucleophilic attack of the resulting anion Rh[P(OCH2CF3)3]4– (2) on the protonated epoxide followed by b-hydride elimination/tautomerization from the resulting rhodium alkyl complex (Scheme 3). Because this mechanism predicts the regeneration of 1, we considered that 1 might catalyze the ring-opening isomerization of styrene oxide. Unfortunately, catalysis was inefficient and produced only ~2 turnovers after 5 days at 55 °C (Table 1).
Table 1. Reaction of 1 with styrene oxide
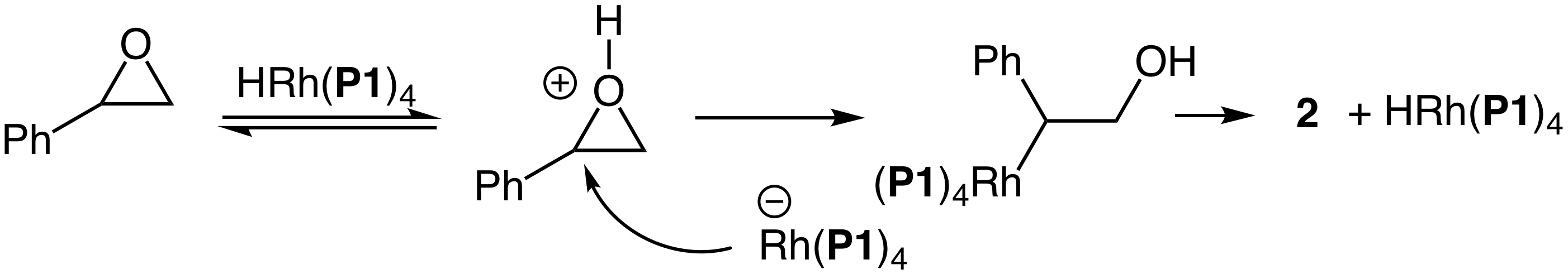
Scheme 3
We considered that the poor reactivity of 1 toward ring-opening isomerization of styrene oxide was due to the low acidity of 1, as our objective was to identify highly acidic group 9 metal hydride complexes for these transformation. To evaluate the acidity of 1, we titrated 1 with triethylamine and determined the equilibrium constant after each addition by 1H and 31P NMR spectroscopy. For example, addition of one equivalent of Et3N to 1 led to formation of a 8:1 mixture of 2:1, which corresponds to an equilibrium constant of 64. From this value, we estimate a pKa value for 1 of 6.7 (Scheme 4).
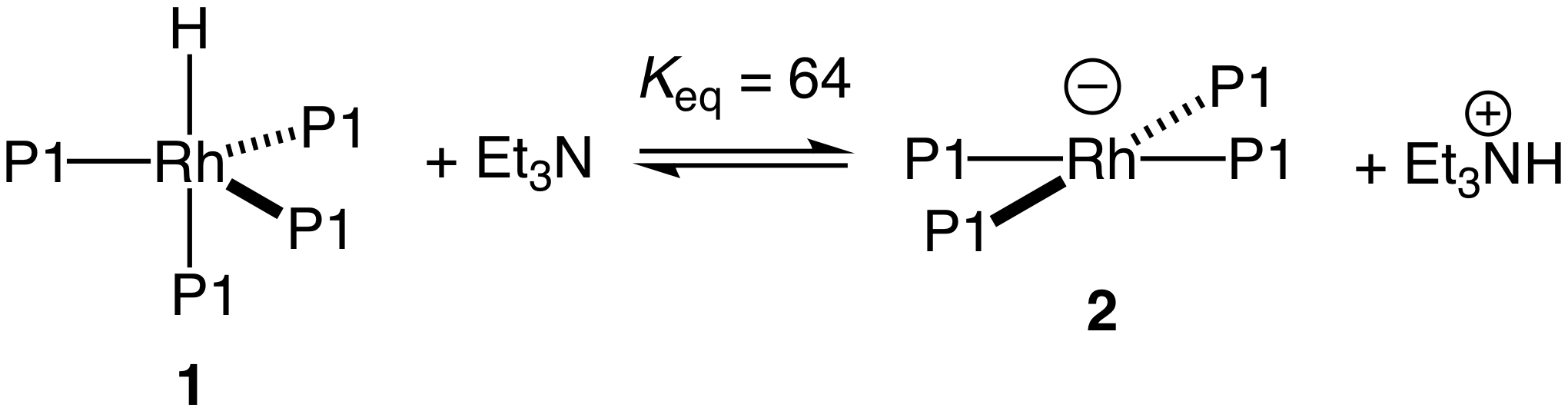
Scheme 4
From the titration experiment depicted in Scheme 4, we conclude that 1 lacks the acidity required to efficiently effect C–O bond cleavage by the mechanism depicted in Scheme 1 and, for this reason, we have focused our efforts on identifying suitably acidic group 9 hydride complexes. Toward this end, we targeted the rhodium phosphine complex HRh{P[OCH(CF3)2]3}4 (4); the P[OCH(CF3)2]3 ligand of 4 is considerably more p-acidic (c = 51) than is the P[OCH2(CF3)]3 ligand of 1 (c = 39). Unfortunately, repeated efforts to synthesize 4 were unsuccessful. Under the assumption that the failure to synthesize 4 was due to the larger cone angle of P[OCH(CF3)2]3 (q = 130°) relative to P[OCH2(CF3)]3 (q = 110°) and the subsequent steric crowding of the resulting tetrakis(phosphite) complex, we targeted the bidentate tetrafluoroaminophosphine ligand 5, which is highly p-acidic owing to the combined effect of the two fluorine atoms and amine group on each phosphine, but which possesses a low steric profile. We have synthesized ligand 5, albeit in low yield from two steps from aniline and PCl3 (Scheme 5).
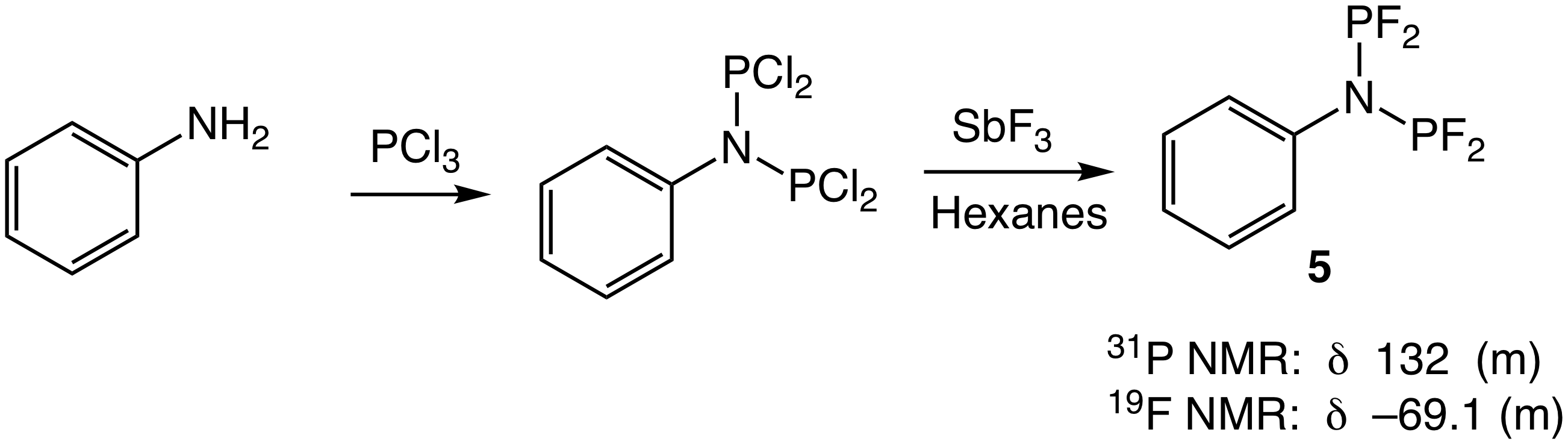
Scheme 5
Future Plans. Our plans in the upcoming funding period are to (1) synthesize rhodium hydride complexes of the form HRh(P–P), where P–P = 5 and related fluorinated bidentate ligands; (2) evaluate the acidity and C–O bond cleavage reactivity of these complexes; and (3) move toward catalytic C–O bond hydrogenolysis in the presence of H2 or HSiR3.










