Reports: UNI156605-UNI1: New Alkene Hydrofunctionalization Reactions: Approaches to Aldehyde Activation Using Base Metal Catalysis
 printer friendly
printer friendlyIntroduction. The purpose of the funded project is to investigate synthetic C–H activation strategies for hydrofunctionalization reactions using first-row transition metal catalysts. These hydrofunctionalization reactions are intended to engage readily available and inexpensive feedstocks originating from petroleum sources such as alkenes, aldehydes and alkynes. The hydrofunctionalization reactions being explored, including hydroacylation, are promising because they can construct new C–C bonds in useful molecules. Moreover, they hold the potential to do so in an inexpensive and atom-economical manner.
This ACS-PRF grant has helped to fund four different undergraduate researchers in my lab, either completely or partially. Students who have worked on these projects have gained knowledge in experimental design, the scientific process, and strategic organic synthetic methodology. They have also gained more specific training in important analytical techniques including nuclear magnetic resonance (NMR) spectroscopy, infrared (IR) spectroscopy, gas chromatography (GC), and mass spectrometry (MS). This ACS-PRF grant has allowed me to pursue my independent research goals and advance my professional career at Samford University. As an assistant professor, scholarship will be an important component of my application portfolio for tenure and promotion. Publication in a peer reviewed journal is a mandatory requirement for promotion and tenure at Samford University. I intend to publish results generated under funding from this ACS-PRF grant within the next two years. Those publications will be indispensable towards my advancement. My progress in research towards these goals is summarized below, including relevant scientific results.
Halogen-Directed Hydroacylation. My ACS-PRF proposal included designs to use the halogens as coordinating directing groups in hydroacylation (Figure 1). Detailed justifications for this approach are found within the original proposal, and include halogen coordination in similar transition metal-catalyzed reactions, and known dative bonding modalities between cobalt and bidentate organohalides. In brief, the majority of transition metal-catalyzed hydroacylation reactions have required coordinative directing action from a heteroatom incorporated in the aldehyde (or less commonly in the alkene) undergoing hydroacylation. While many common coordinative directing groups such as phenols and sulfides have already been demonstrated as useful functional handles post-hydroacylation, organohalides are some of the most versatile and easily manipulated compounds available. Using the halogens as directing groups for hydroacylation would make more elaborate bond construction accessible because the halogen atom would switch seamlessly from a necessary directing group to an advantageous functional group.
We began our studies by examining hydroacylation with 2-bromobenzaldehyde in combination with several different alkynes and alkenes. My research program currently involves several of the first row transition metals including Fe, Co, Ni and Cu. Our initial attempts involved Co since there have been several reports of hydroacylation using this base metal as a catalyst. Many of the previous Co-catalyzed hydroacylation reactions have used air-stable Co(II) precatalysts with added reductants as activators. This obviates the need to synthesize and handle Co(I) complexes which are typically air and moisture sensitive. We chose a similar approach and tested Co(II) precatalysts such as CoCl2 and (PPh3)2CoCl2 with Zn, In or Mn as activators. Monodentate and bidentate phosphines were examined because there was precedent for them serving as ancillary ligands in both Rh and Co-catalyzed hydroacylation and because they should allow for an open coordination site at the metal center (to allow the directing group to exert its influence). Variables such as precatalyst, ancillary ligand, reductant, solvent, temperature and time were most frequently tested on the alkyne phenylacetylene (Figure 2), although other unsaturated compounds were also tested (not shown). Reactions were analyzed via thin layer chromatography (TLC), NMR, and GC-MS.
Tandem Hydroacylation-Heck Reactions. The halogen-directed Co-catalyzed hydroacylation reactions described above proved recalcitrant, with only trace product formation observed. Despite there being ample precedent for orthogonal reactivity with Co catalysis (detailed in the original proposal), unproductive conversion of the starting material was high and reactivity of the C–Br bond under these conditions was evident. Longer reaction times appeared to cause product decomposition and byproduct formation. My research group is still investigating reactions similar to those described in the previous section, but our efforts have also focused on using the inherent reactivity of the halogen group within similar tandem reactions. This aligns with the original intent of the proposal since the ultimate goal when considering halogen atoms as directing groups is to employ them within subsequent C–C bond forming reactions. While Rh and Co have dominated reports of intermolecular hydroacylation, Ni complexes have also been reported as a catalysts for this important reaction. More importantly, Ni complexes are known to catalyze hydroacylation and C–X activating reactions such as the Heck coupling under similar conditions. My group proposed a tandem hydroacylation-Heck reaction, and we began our investigation with 2-bromobenzaldehyde and several symmetrical alkynes. We chose Ni(COD)2 as a precatalyst and tricyclohexylphosphine (P(Cy)3) as a supporting ligand since this combination had recently been reported effective for hydroacylation. Similar conditions with Ni complexes also catalyze the Heck reaction with aryl halides so long as a base is present. We chose potassium phosphate (K3PO4) as the base in our initial attempts (Figure 3). Subsequent optimization showed that we could use an air-stable Ni(II) precatalyst with a chelating 1,2-bis(diphenylphosphino)ethane ligand ((DPPE)NiCl2). The precatalyst could be reduced to a catalytic species using Zn, although activation times are long (around 3 hours for optimal yield). Optimization also showed that the soluble organic base triethylamine was more effective. The reaction can yield up to 70% product (by GC analysis against an internal standard).
Our current efforts are focused on continuing optimization of the tandem hydroacylation-Heck reaction. Higher chemical yields would be more appropriate for publication. The long activation times also make the reaction conditions somewhat cumbersome. Efforts to explore pre-activated Ni catalysts are underway for these reasons. Demonstrating a high functional group compatibility will also aid in the publication of these results. We are still currently investigating nontraditional directing groups in Co-catalyzed hydroacylation as delineated in the original proposal.
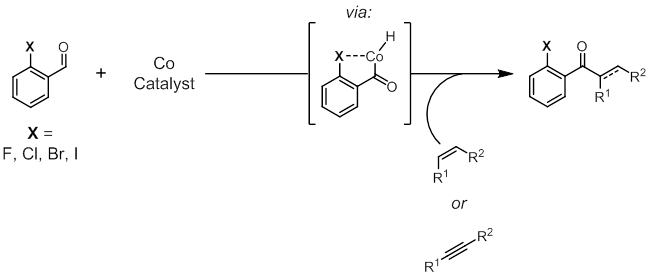
Figure 1
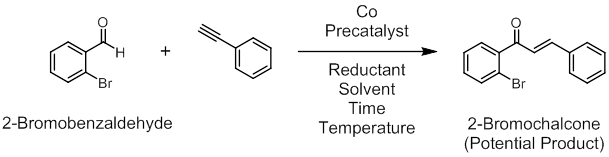
Figure 2
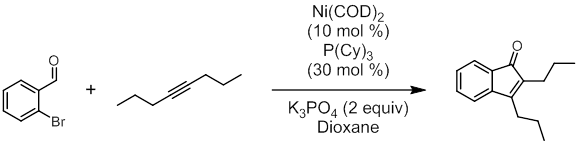
Figure 3
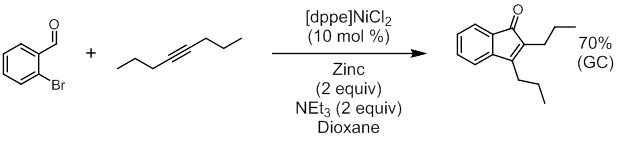
Figure 4










