Reports: UR455266-UR4: The Use of a Cyclopropylcarbinyl Radical Rearrangement as a Singlet Diradical Probe
 printer friendly
printer friendlyBicyclo[3.2.0]hept-2-ene (1) undergoes thermal isomerization to norbornene (2) in the gas phase (Scheme 1). This rearrangement has served as a historical exemplar of a [1,3] sigmatropic carbon migration. Competitive isomerization and fragmentation processes at temperatures in excess of 300 ¼C convert 1 to its isomer 2 or to fragments cyclopentadiene and ethylene (Scheme 1).1,2 Despite the large experimental error in activation parameters for the [1,3] process, Cock and Frey conclude that the activation parameters for the sum (k13 + kf) are close to the expected values "if both processes have a biradical mechanism" and therefore that "a two-step pathway is competitive."
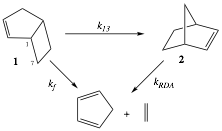
Scheme 1. Thermal Reactions of Bicyclo[3.2.0]hept-2-ene (1)
In a dramatic reversal of this mechanistic rationale, Woodward and Hoffmann declared emphatically that exo-7-d-endo-6-acetoxybicyclo[3.2.0]hept-2-ene "undergoes a concerted symmetry-allowed suprafacial [1,3] shift, with inversion at the migrating center, É at 307 ¼C."3 Due to the steric inhibition of antarafacial migration in bicyclo[3.2.0]hept-2-enes, only suprafacial migration is geometrically feasible. According to the Woodward-Hoffmann rules, suprafacial migration with inversion of configuration (si) is privileged as orbital-symmetry allowed and suprafacial migration with retention of configuration (sr) is orbital-symmetry forbidden. As more experimental evidence has been compiled for negligible stereoselectivity, by examining the more conformationally flexible bicyclo[4.2.0]oct-2-enes, the mechanistic viewpoint has gained prominence that [1,3] sigmatropic rearrangements traverse short-lived diradical transition structures residing on a shallow energy surface.7 The mixed stereochemical results that have often converged toward a value of unity have effectively subverted alternative mechanistic interpretations such as competing orbital symmetry-allowed (si) and orbital symmetry-forbidden (sr) pathways8 or competing concerted (si) and diradical (si/sr) mechanisms.9 The potential however for short-lived nonequilibrated diradical intermediates partitioning between inversion and retention modes10 deserves more critical examination.
It has been 15 years since our report on the thermal rearrangement of exo-7-methylbicyclo[3.2.0]hept-2-ene (3) in the gas phase at 275 ¼C (Scheme 2).2 We proposed to examine our earlier assumption that all exo-7-alkylbicyclo[3.2.0]hept-2-enes would yield comparable results both kinetically and stereochemically.
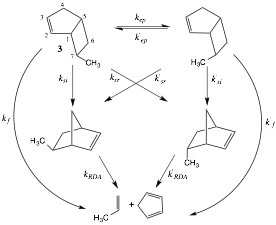
Scheme 2. Thermal Chemistry of exo-7-Methylbicyclo[3.2.0]hept-2-ene (3)
The rate constants for the overall rate of decomposition (kd) for a series of exo-7-alkylbicyclo[3.2.0]hept-2-enes are listed in Table 1 from most reactive exo-7-propylbicyclo[3.2.0]hept-2-ene to the least reactive exo-7-isopropylbicyclo[3.2.0]hept-2-ene. Given that all of the exo-7-alkylbicyclo[3.2.0]hept-2-enes afford a secondary alkyl-allyl diradical upon homolytic cleavage of the C1-C7 bond (Scheme 3), it is not surprising that the kd values for the most reactive entry is less than a factor of two greater than the least reactive entry. The relative order of importance of kinetic processes is k13 > kf in all cases but the k13/kf ratios are much lower for all entries compared with exo-7-methylbicyclo[3.2.0]hept-2-ene (3) (Table 1).
The rates of retro Diels-Alder reactions for all exo-5-alkylnorbornenes were comparable as were those for the endo-5-alkylnorbornenes. In all cases the reactivity of the endo-5-alkylnorbornenes exceeded that of the corresponding exo-5-alkylnorbornenes by a factor greater than 2 (Table 2).
The absence of a global trend in how the resultant non-equilibrated singlet diradical bifurcate between rearrangement and fragmentation (Scheme 3) suggests multiple operative factors once the initial transition structure is reached and the intermediate begins to sample the available conformational space. Examining the % contributions of the si and sr products directly (Table 3) reveals that the si product is highly favored, consistent with the predictions of Carpenter11 and Houk12. The % si range varies narrowly from 80-89%, an outcome consistent with conservation of angular momentum or preservation of the moment of inertia once the endo trajectory12 commences.
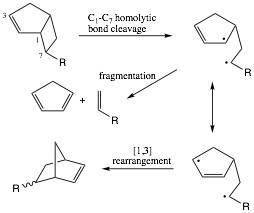
Scheme 3. Bifurcation of 2¼ Alkyl-Allyl Diradical
between Rearrangement and Fragmentation
Table 1. Rate Constants and Exit Channels for
exo-7-Alkylbicyclo[3.2.0]hept-2-enes
R-
| kd @ 275 ¼C
| krel | k13/kf | % [1,3]
| % epim
| % frag
|
Pr- | 2.0 « 10-5 s-1 | 1.8 | 3.2 | 76 | 0 | 24 |
Bu- | 1.9 « 10-5 s-1 | 1.7 | 1.8 | 64 | 0 | 36 |
Et- | 1.6 « 10-5 s-1 | 1.5 | 5.4 | 84 | 0 | 16 |
Me- | 1.5 « 10-5 s-1 | 1.4 | 150 | 95 | 4 | 0.6 |
t-Bu- | 1.4 « 10-5 s-1 | 1.3 | 2.9 | 74 | 0 | 26 |
i-Pr- | 1.1 « 10-5 s-1 | 1.0 | 1.9 | 65 | 0 | 35 |
Table 2. Rates of Retro Diels-Alder Reactions for
exo- (kRDA) and endo- (k¢RDA)-7-Alkylbicyclo[3.2.0]hept-2-enes
R-
| kRDA | k¢RDA
| k¢RDA /kRDA
|
Et- | 2.5 « 10-4 s-1 | 5.5 « 10-4 s-1 | 2.2 |
Me- | 2.0 « 10-4 s-1
| 4.7 « 10-4 s-1
| 2.3 |
Pr- | 2.5 « 10-4 s-1
| 6.0 « 10-4 s-1
| 2.4 |
i-Pr- | 1.7 « 10-4 s-1
| 4.6 « 10-4 s-1
| 2.7 |
Bu- | 1.8 « 10-4 s-1
| 5.0 « 10-4 s-1
| 2.8
|
t-Bu- | 9.4 « 10-5 s-1
| 6.1 « 10-4 s-1
| 6.5
|
Table 3. Stereoselectivity of [1,3] Carbon Shifts in
exo-7-Alkylbicyclo[3.2.0]hept-2-enes
R-
| ksi | ksr
| si/sr
| %si
| %sr
|
Et- | 1.2 « 10-5 s-1 | 1.5 « 10-6 s-1 | 8.0 | 89 | 11 |
i-Pr- | 6.5 « 10-6 s-1 | 8.1 « 10-7 s-1 | 8.0 | 89 | 11 |
Me- | 1.2 « 10-5 s-1
| 1.9 « 10-6 s-1
| 6.3 | 86 | 14 |
Pr- | 1.3 « 10-5 s-1
| 2.2 « 10-6 s-1
| 5.9 | 86 | 14 |
t-Bu- | 8.3 « 10-6 s-1
| 2.1 « 10-6 s-1
| 4.0 | 80 | 20 |
Bu- | 9.7 « 10-6 s-1
| 2.5 « 10-6 s-1
| 3.9
| 80 | 20 |
[1] H. M. Frey, A. T. Cocks, J. Chem. Soc. A 1971, 2564-2566.
[2] J. D. Bender, P. A. Leber, R. R. Lirio, R. S. Smith, J. Org. Chem. 2000, 65, 5396-5402.
[3] R. B. Woodward, R. Hoffmann, The Conservation of Orbital Symmetry, Verlag Chemie: Weinheim, 1970, pp 119-122.
[4] J. J. Gajewski, Acc. Chem. Res. 1980, 13, 142-148.
[5] J. A. Berson, G. L. Nelson, J. Am. Chem. Soc. 1967, 89, 5503-5504.
[6] P. A. Leber, J. E. Baldwin, Acc. Chem. Res. 2002, 35, 279-287.
[7] J. E. Baldwin, P. A. Leber, Org. Biomol. Chem. 2008, 6, 36-47.
[8] J. A. Berson, Acc. Chem. Res. 1972, 5, 406-414.
[9] G.-G. KlŠrner, R. Drewes, D. Hasselmann, J. Am. Chem. Soc. 1988, 110, 297-298.
[10] J. E. Baldwin, K. D. Belfield, J. Am. Chem. Soc. 1988, 110, 296-297.
[11] B. K. Carpenter, J. Am. Chem. Soc. 1995, 117, 6336-6344.
[12] S. Wilsey, K. N. Houk, A. H. Zewail, J. Am. Chem. Soc. 1999, 121, 5772-5786.










