Reports: DNI355327-DNI3: Incorporating Lewis Acid Anion Co-Catalysts into Homogeneous Transition-Metal Systems for Carbon-Carbon Bond Formation
 printer friendly
printer friendlyMuch societal value has resulted from the design of new organometallic catalysts, a practice that demands precise control over catalyst structure and function, as well as a mechanistic understanding of the catalytic process. The research project supported by the Petroleum Research Fund looks beyond conventional catalyst design parameters (metal and ligand) to outer sphere (or secondary coordination sphere) influences, in the pursuit of more selective and useful catalytic reactions. Within that wider effort, my group is working to study how synthetic catalysts in solution interact with the reaction environment, and how these interactions influence reaction outcomes.
Our proposal focused on bimetallic ion pair scaffolds - composed of two oppositely charged metal complexes - for small molecule synthesis. To achieve our long-term goal of a general bimetallic catalyst platform, we are investigating how metallic additives boost selectivity and reactivity in existing catalytic systems, and mapping the synthetic approaches and catalytic applications of bimetallic ion pairs. These initial stoichiometric demonstrations will direct our future catalyst development, such as methods for cross-coupling and hydroesterification. In addition to discovering improved catalytic processes, the study of ion pairing phenomena in organic solutions will have broader significance in many areas of reaction development. Funding from PRF has enabled significant progress towards our lab’s long-term aims, and has aided the professional development of multiple graduate students, an undergraduate and a postdoctoral scholar.
Accelerating organometallic reactions with metallic co-catalysts. Lewis acidic additives have been widely used to accelerate several classes of challenging transition metal-mediated reactions, such as the insertion of carbon monoxide into M-R bonds, a very common elementary step in organometallic reactions. Recently we have shown the Pd/xantphos-catalyzed cross coupling of amides and aryl halides is accelerated by co-catalytic metal triflate additives, improving yields for a variety of N-aryl amide products when metal triflates (M(OTf)3) are employed as a catalytic additive. The observation of an aryl halide dependence on rate and other kinetic experiments are consistent with a mechanism in which ligand exchange of halide for amide (“transmetalation”) is turnover limiting. Based on kinetic data and stoichiometric reactions of putative catalyst intermediates, we propose the accelerating effect originates from a reversible Lewis acid-mediated halide abstraction during catalysis (Scheme 1).
![]()
Scheme 1. M(OTf)3 halide abstraction in a Pd-catalyzed amide N-arylation.
With an improved Pd/xantphos catalyst system for N-arylation of amides in hand, we are now expanding our synthetic exploration with the end goal of demonstrating the value of our protocol in specific synthetic organic applications (secondary carboxylic amides and sulfonamides) and partnering with industrial collaborators to more rapidly evaluate co-catalysts and conditions in high-throughput screens.
Joining together oppositely charged organometallic complexes to generate stable heterobimetallic ion pairs. We are currently preparing bimetallic ion pairs to exert the influence of exogenous additives on a catalytic reaction while leveraging the power of effective molarity to enforce high proximity between catalyst and additive. Our first efforts resulted in organometallic anions based on an anionic N-heterocyclic carbene (ANHC) ligand, which we previously employed in a zwitterionic alkyne hydration precatalyst (ANHC)Au(SMe2). We have paired anions [(ANHC)Au(OAr)]- with Pd complexes with known catalytic relevance and are exploring the potential of bimetallic catalysts for use in cross-coupling and in carbonylation (e.g., using carbon monoxide for the synthesis of esters and amides). The Au/Pt ion pair in Figure 1 reacts with CO to generate the corresponding ester product, our first successful demonstration of a productive bond formation driven by the joint reaction of two oppositely charged ionic complexes.
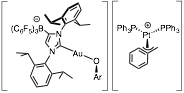
Figure 1. A Au/Pt bimetallic ion pair.
We are now working to build off our initial findings to implement Au/Pt and Au/Pd-based ion pairs in catalytic reactions. Our group will continue to find routes towards rational catalyst design through understanding (and exploiting) interactions in the secondary coordination sphere – both through examining the effects of exogenous additives and by designing de novo structures capable of enforcing intermolecular association.










