Reports: DNI656191-DNI6: A Computational Approach for Optimizing Metal-Free Synthesis of Conjugated Polymers
 printer friendly
printer friendlyThis project aims to establish reaction pathways for the polymerization of b-silyl ketenes, SiR3(H)C=C=O. Due to their C=C/C=O bifunctionality, ketenes can polymerize to yield various, previously unrealized architectures, such as poly(silyl ketone) and poly(silyl acetal) [Fig. 1]. Consequently, this project has the potential to facilitate the creation of polymers with new chemical compositions from fossil fuel sources. Moreover, the comparatively high reactivity of ketenes allows for uncatalyzed polymerization, thus having potential to facilitate metal-free polymerization. This could remove costly steps such as product purification.
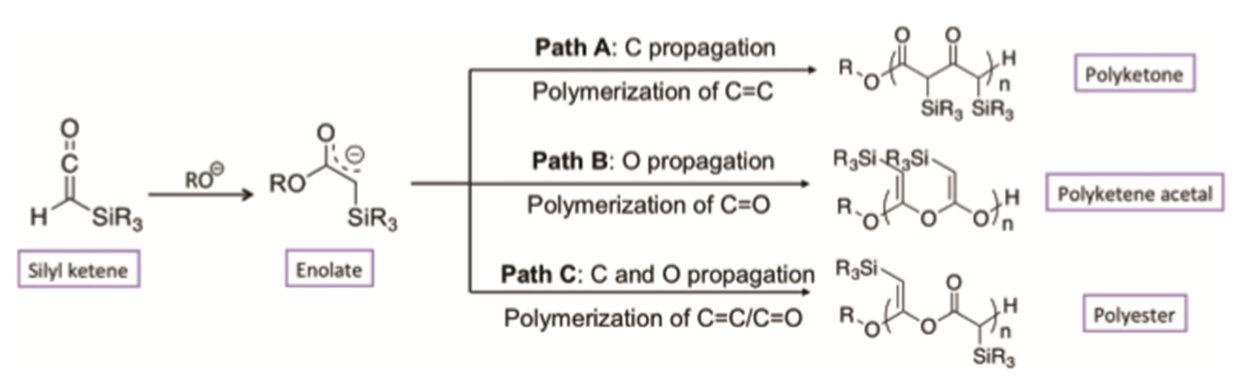
Figure 1: Proposed anionic polymerization of silyl ketenes with alkoxides. Initiation occurs via nucleophilic attack on Ca, yielding an enolate that can propagate both via the O- and Cb- functionality. Propagation continues by nucleophilic addition to the Ca on the next silyl ketene monomer, which in turn forms a reactive enolate, etc. Reaction paths A-C illustrate three limiting cases of possible propagation pathways.
Milestones achieved during the reporting period include demonstrating the feasibility of b-silyl ketene polymerization and identifying factors that influence the chemical composition of the polymerization product. More specifically, we evaluated the feasibility of controlled polymerization via radical and anionic polymerization, and identified reaction conditions to influence the reaction rates and thermodynamically preferred polymerization products. This work was performed through an in-kind cooperation with our experimental collaborator (E. Pentzer, Case Western University). We established computational protocols that balance accuracy and computational efficiency to facilitate predictive calculations of reaction pathways, activation barriers, and reaction energies while efficiently tackling large numbers of conformations of the flexible polymer chain. We found that an accurate conformational sampling is demanding on the computational approach, because it requires a balanced description between steric repulsion and attractive dispersion interactions of the typically bulky silyl residues. We employed semiempirical predictions (PM6 and PM7) to explore conformations and reaction paths using the ZStruct transition state optimizer. Structures within a typically 2-4 kcal/mol energy window were further refined using density functional theory (up to wB97X-D/6-311G(3df,2pd) level), which we found to reproduce CCSD(T)/cc-pVTZ benchmarks to within ~1 kcal/mol.
Key findings and results for the reporting period are:
1. b-silyl ketenes readily undergo anionic polymerization with alkoxide initiators.
We confirmed our underlying hypothesis that b-silyl ketenes are sufficiently reactive to undergo anionic polymerization with anionic alkoxide initiators. More specifically, initial polymerization experiments show that anionic polymerization of triisopropyl-silyl (TIPS) ketene with tertiary butyl oxide (tBuO-) yields products with molecular weights up to 12,000 Dalton at up to 94% conversion. Chemical compositions of the products are consistent with the polymerization of C=C and C=O functionalities (see below). This finding should be contrasted with results for radical polymerization, which leads to early termination of the polymerization under all circumstances tested. Early termination of the radical propagation is caused by several side reactions, such as dimerization of the initiating species, as found e.g. for azobisisobutyronitrile (AIBN), or formation of unreactive cyclic oligomers, as found e.g. for alkoxy radical initiators. Anionic polymerization is therefore the preferable polymerization route.
2. Propagation can occur both via the C=C and C=O functionality.
Polymerizing triisopropyl-silyl (TIPS) ketene in tetrahydrofurane at low temperatures yields products with mixed chemical functionalities. Nuclear magnetic resonance and infrared characterization indicate a mixture of ketone, ester, acetal, and olefin functionalities [Fig. 2]. These assignments were confirmed from computed infrared vibrational frequencies for structural candidates.
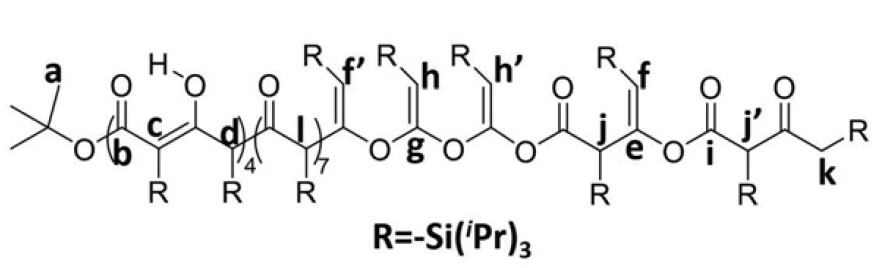
Figure 2: Representative composition of an oligomer obtained from reacting TIPS ketene with tBuO-, as assigned based on 13C NMR.
3. Solvent polarity and counterions influence the activity and preference for propagation via the C=C and C=O bonds.
The chemical composition of the polymerization product depends on several factors. Experiment and theory agree that the identity of the counterion (from the anionic initiator) influences the reaction rate and chemical composition of the product. For example, our computational predictions suggest that polymerizing TIPS ketene in the presence of Na+ counterions leads to a thermodynamic preference of propagation via the C- atom of the enolate intermediate, while K+ promotes alternating between propagating via C- and O- [Fig. 3]. Additionally, the calculated apparent activation barrier for K+ is estimated to be ~55% lower than for Na+, leading to faster polymerization in the presence of potassium counterions. These results agree qualitatively with experiment, which finds barriers of ~16 and ~13 kcal/mol in the presence of sodium and potassium, respectively. A qualitative explanation for these differences is that the smaller sodium ions associate more effectively with the propagating enolate, thus hindering propagation to a certain degree. The larger potassium ions, on the other hand, are more loosely associated with the propagation site because of steric demands of the TIPS residues, and therefore allow for faster propagation. Interestingly, the different counterions stabilize the negative charge on different centers, thus influencing the propagation route. Moreover, our calculations predict that solvent polarity also influences reaction energies and activation barriers by differently stabilizing negative charges on the C-/O- sites of the enolate [Fig. 4]. Going forward, we aim to exploit these counterion/solvent effects to create tailored “nano-solvation” environments that will control the formation of products with specific chemical compositions.
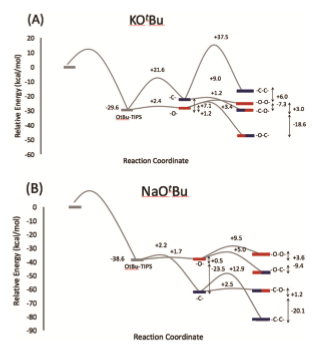
Figure 3: Our computations predict that the nature of the counterion controls the energetic ordering of products, i.e., the thermodynamics of the polymerization, as well as the relative activation barriers, i.e., the polymerization kinetics. The notation “-X-Y-“ denotes a trimer formed by propagating via the X- functionality in the first step and the Y- functionality in the subsequent step. Importantly, different choices of counterions allow one to tilt the balance between alternating (“-O-C-“) and uniform (“-O-O-” and “-C-C-“) propagation.
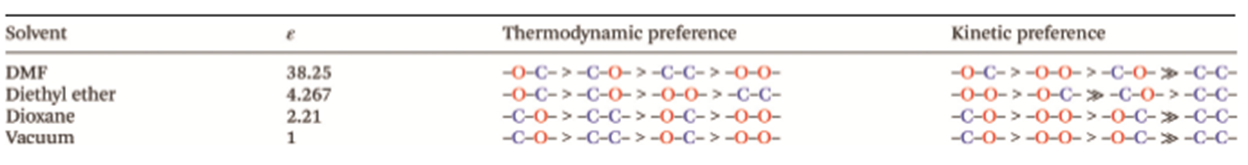
Figure 4: Our computations predict that the solvent polarity controls the energetic ordering of products, i.e., the thermodynamics of the polymerization, as well as the relative activation barriers, i.e., the polymerization kinetics. Interestingly, the most significant change occurs in a relatively small window for the dielectric constant, i.e. between e = 2.2 and 4.3.










