Reports: DNI355302-DNI3: Mechanism and Scope of Bis(imino)pyridine Manganese-Catalyzed Hydrosilylation
 printer friendly
printer friendlyOverview. During this project, we have investigated several aspects of bis(imino)pyridine (or pyridine diimine, PDI) manganese-mediated hydrosilylation. Notably, we have elucidated two competing, yet complimentary, mechanisms through which hydrosilylation takes place and have expanded the scope to include a broad range of aldehyde-, ketone-, formate-, and ester-containing substrates. These aspects are described below along with the overall impact of this research.
Mechanistic Observations. In 2014, we reported that (Ph2PPrPDI)Mn (1) (Figure 1) exhibits activity for the hydrosilylation of ketones and dihydrosilylation of esters. Under stoichiometric and catalytic conditions, we found that adding PhSiH3 to (Ph2PPrPDI)Mn results in partial conversion to a diamagnetic hydride complex that is believed to feature a silylated chelate. To evaluate the catalytic competency of this species, (Ph2PPrPDI)MnH (2) (Figure 2) was independently prepared by adding excess NaEt3BH to (Ph2PPrPDI)MnCl2. Kinetics experiments were then conducted to gauge the efficacy of 1 and 2 in carbonyl and carboxylate hydrosilylation reactions. By following the disappearance of diisopropyl ketone, we determined that 1 is more active than 2 for carbonyl hydrosilylation and that this transformation is first order with respect to catalyst, substrate, and PhSiH3. To our surprise, monitoring the disappearance of isopropyl formate revealed that 2 is considerably more active than 1 for carboxylate dihydrosilylation. Moreover, a kinetic isotope effect (KIE) of 2.2 ± 0.1 was observed for the 1-catalyzed hydrosilylation of diisopropyl ketone, while the 2-catalyzed reaction revealed a KIE of 4.1 ± 0.6, suggesting two different mechanisms of hydrosilylation.
Modified Ojima Mechanism. Considering our observations, we proposed that 1 achieves carbonyl and carboxylate hydrosilylation through the modified Ojima mechanism in Figure 1. The initial step involves phosphine dissociation and Si-H oxidative addition to generate a 5-coordinate silyl hydride intermediate. Upon substrate coordination, insertion into Mn-H is believed to be the rate-determining step, consistent with a first order dependence on both substrate and silane. Once formed, the resulting silyl alkoxide undergoes reductive elimination to yield the silyl ether product. For carboxylates, beta-alkoxide elimination occurs before reductive elimination to eliminate aldehyde, which may re-enter the catalytic cycle. Throughout this process, it is believed that Mn remains divalent due to the redox-activity of Ph2PPrPDI.
 |
Figure 1. Modified Ojima mechanism for 1-catalyzed carbonyl and carboxylate hydrosilylation.
Hydride Insertion Mechanism. Since hydride species analogous to 2 are formed during hydrosilylation, it was also proposed that a direct insertion mechanism (Figure 2) contributes to the observed catalysis. Phosphine displacement by incoming substrate allows for insertion into the Mn-H moiety of 2, generating an alkoxide intermediate. This intermediate may then undergo rate-determining sigma-bond metathesis with silane to yield silyl ether and generate 2, or in the case of carboxylates, undergo fast beta-alkoxide elimination. Although the electronic structure of 2 is consistent with a Mn(III) center supported by a PDI dianion, Mn is believed to remain divalent throughout the remainder of the cycle due to the redox flexibility of the chelate.
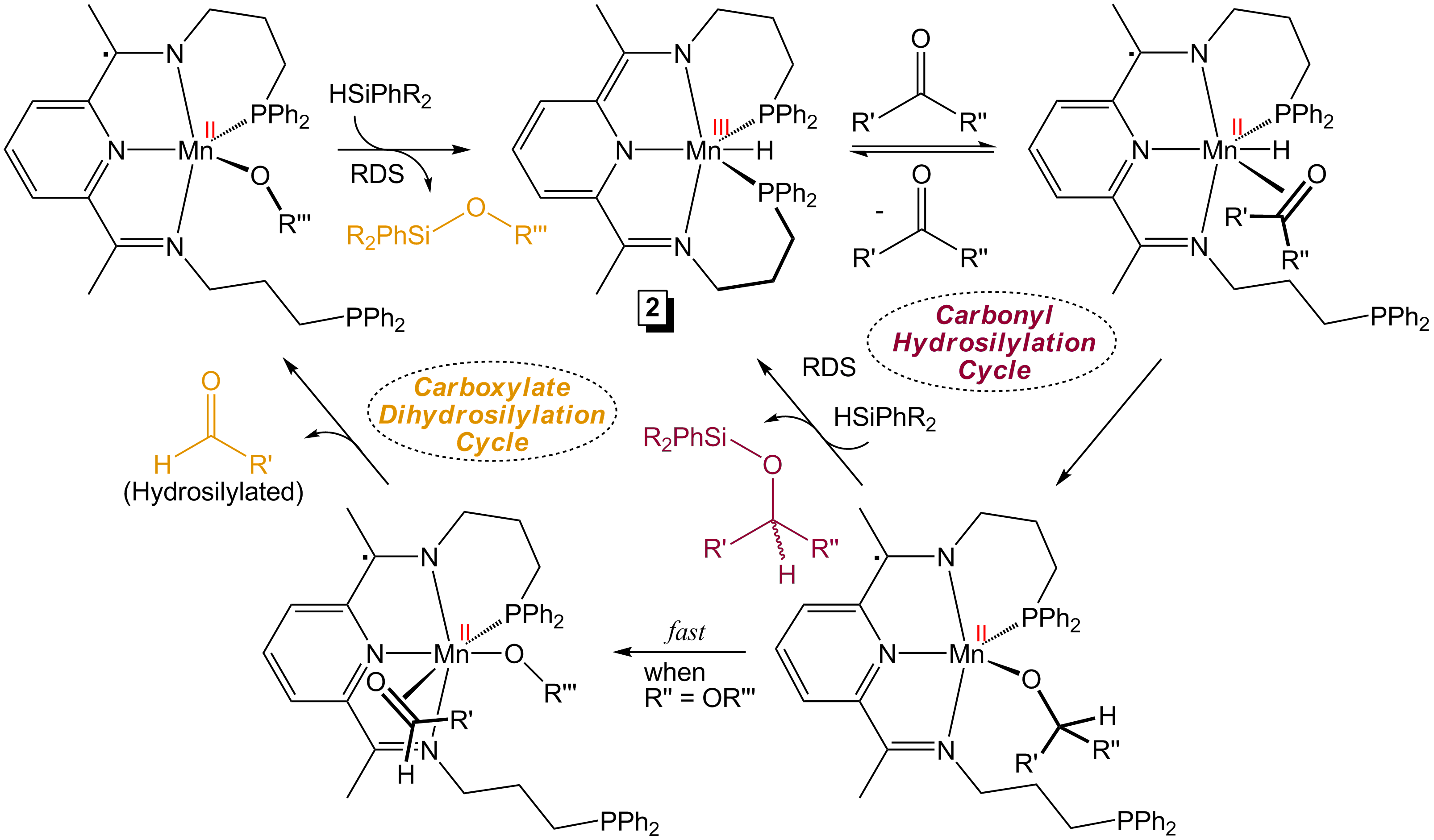
Figure 2. Insertion mechanism for 2-catalyzed carbonyl and carboxylate hydrosilylation.
Carbonyl Hydrosilylation Scope. We have also demonstrated an expanded scope for 1-mediated hydrosilylation. Adding a neat mixture of benzaldehyde and PhSiH3 to 0.1 mol% 1 at 25 °C resulted in complete conversion to silyl ethers within 2 min and treatment with 10% aq. NaOH allows for isolation of benzyl alcohol following extraction. Thirteen different aldehydes have been hydrosilylated under these conditions and 1 has been found to tolerate a range of functionalities. For unsaturated aldehydes such as 3-cyclohexene-1-carboxaldehyde and citral, the aldehyde functionality is reduced and the olefin is not, consistent with our previous attempts at (PDI)Mn alkene hydrosilylation. Additionally, the hydrosilylation of benzaldehyde, 4-fluorobenzaldehyde, 2-naphthaldehyde, and furfural has been achieved using 0.01 mol% 1 under identical conditions to yield the respective silyl ethers with TOFs of 4,900 min-1. To determine maximum TON for 1-catalyzed aldehyde hydrosilylation, a mixture of 10,000 equivalents of PhSiH3 and benzaldehyde was added to 1 and repeated 4 times (total of 50,000 equivalents) at 15 min intervals. Analysis of the resulting mixture revealed 67% conversion, indicating a maximum TON of 31,000.
Carboxylate Hydrosilylation Scope. In 2014, we reported that 1 exhibits modest activity for the dihydrosilylation of esters and have recently expanded the scope of this reaction to include formates. When a neat equimolar mixture of methylformate (or ethylformate) and PhSiH3 is added to 0.02 mol% 1, an exothermic reaction occurs with >99% substrate conversion in 15 min. Attempts to hydrolyze the resulting silyl ethers to isolate the corresponding alcohols by distillation did not allowed for adequate separation, so higher molecular weight formates were screened. Adding an equimolar quantity of benzyl formate and PhSiH3 to 0.02 mol% of 1 or 2 results in vigorous bubbling. Catalyst deactivation after 15 min revealed complete consumption of formate. Hydrolysis using 10% aq. NaOH, followed by extraction and evaporation, afforded pure benzyl alcohol in excellent yield (88%). Similarly, p-anisyl formate, phenylethyl formate, hexyl formate, and isoamyl formate were reduced to the corresponding alcohol. A TOF of 330 min-1 (based on substrate conversion) was achieved for each substrate, which is the highest reported TOF for transition metal catalyzed carboxylate dihydrosilylation.
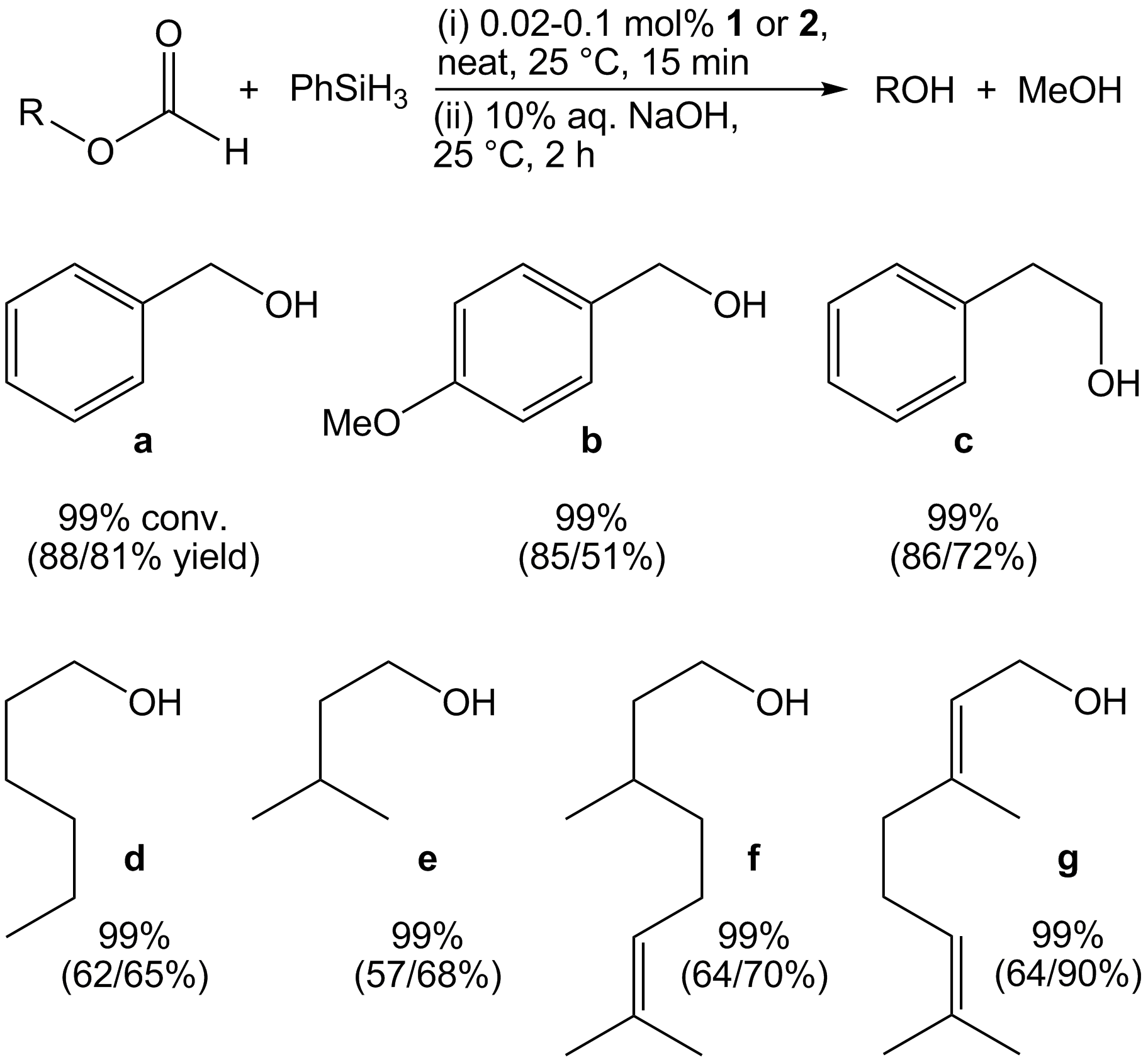
Figure 3. Optimization of 1- and 2-catalyzed formate dihydrosilylation.
Project Impact. Since our initial communication of 1-mediated hydrosilylation, a wave of publications describing manganese catalysis has inundated leading chemistry journals. It has become obvious to us and the broader field that manganese catalysts were considerably overlooked prior to 2014. Our efforts have also inspired several publications involving manganese-catalyzed hydrogenation, asymmetric carbonyl hydrosilylation, and even silicone cross-linking. Given its effectiveness, compound 1 was recently made commercially available by Sigma-Aldrich Corporation (a subsidiary of Merck KGaA). Our research group also received an NSF CAREER Award to evaluate the role of ligand structure on hydrosilylation activity and the ability of manganese catalysts to prepare silicones via hydrosilylation and the dehydrogenative silylation of alcohols. A graduate student who worked on this project earned his Ph.D. and went on to conduct postdoctoral studies at the University of Michigan. An undergraduate involved with this project earned a B.S. degree and is now employed by B&W Tek.










