Reports: DNI654240-DNI6: Mesoscale Simulation and Machine Learning of Asphaltene Aggregation
 printer friendly
printer friendlySynopsis. Building on our results from the previous two years, over the past 12 months of this award we have made four principal achievements.
First, we have analyzed of the morphological behavior of asphaltene aggregation at a variety of temperatures, pressures, and n-heptane/toluene solvent compositions using nonlinear manifold learning techniques to construct low-dimensional assembly landscapes. These landscapes have shed new molecular-level understanding of aggregation behavior. In particular, they revealed an interesting inversion of the effect of pressure upon aggregation depending on the solvent composition – elevated pressure suppresses aggregation in n-heptane, but enhances aggregation in toluene. They have also shown the effect of increasing temperature and increasing toluene concentration to modulate the molecular assembly landscape in very similar manners –stabilization of small clusters and loose cluster configurations. This has led us to the finding that toluene – and perhaps asphaltene dispersants more generally – can be considered to act as an “effective temperature” in suppressing aggregation.
Second, we have extended our protocol for the construction of coarse-grained models of asphaltene molecules from all-atom simulation data to archipelago asphaltene architectures. This now enables us to efficiently generate coarse-grained molecular models of essentially any asphaltene molecule.
Third, we have performed coarse-grained molecular simulations of the aggregation of three prototypical archipelago asphaltenes, characterized the aggregation morphologies, and measured the fractal dimensions of the networks formed at high concentration. We find that the aggregation behavior – as we previously showed for continental architectures – is in line with the Yen-Mullins hierarchy, but the details depend strongly on the specifics of the molecular architecture. For example, molecules with more cores tend to aggregate more readily, and those with linear topologies tend to form fractal networks more efficiently than those which exist in cyclic forms.
Fourth, we have applied manifold learning to systematically construct low-dimensional folding landscapes for three prototypical archipelago asphaltenes. By computing free energy landscapes supported by these low-dimensional landscapes, we have resolved the diversity metastable states populated by these molecules in thermodynamic equilibrium to reveal a rich and complex morphological behavior reminiscent of protein folding.
Inversion
in the pressure dependence of asphaltene aggregation. In pure n-heptane
solvent, increasing pressure serves to destabilize large asphaltene clusters
and suppress aggregation (Fig. 1a). This is consistent with a positive
volume change of assembly ![]() ,
where ΔG and ΔV are the change in free energy and volume for assembly. Conversely,
in pure toluene solvent, elevated pressure enhances aggregation (Fig.
1b), consistent with a negative ΔV. To the best of our knowledge,
this interesting inversion in the volume change of assembly as a function of
solvent composition has not been previously reported.
,
where ΔG and ΔV are the change in free energy and volume for assembly. Conversely,
in pure toluene solvent, elevated pressure enhances aggregation (Fig.
1b), consistent with a negative ΔV. To the best of our knowledge,
this interesting inversion in the volume change of assembly as a function of
solvent composition has not been previously reported.
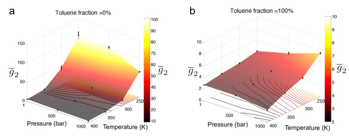
Fig. 1 – Inversion in effect of pressure upon aggregation as a function of solvent composition for a prototypical continental asphaltene. g2 is the mass-averaged cluster size.
Toluene acts as an effective temperature. Nonlinear manifold learning applied to coarse-grained molecular simulations of the aggregation of a prototypical continental asphaltene at a variety of temperatures, pressures, and solvent compositions discovered a single collective order parameter Λ in which to construct pseudo-1D assembly landscapes. These landscapes map out the relative stability of various asphaltene aggregates in thermodynamic equilibrium, and provide a quantitative measure of the effect of manipulating external conditions on the morphological distribution. We find that increasing toluene solvent fraction and increasing temperature modulate the landscapes in a similar fashion. This indicates that although the underlying mechanisms differ, toluene can be considered to act upon asphaltene assembly as an “effective temperature”.
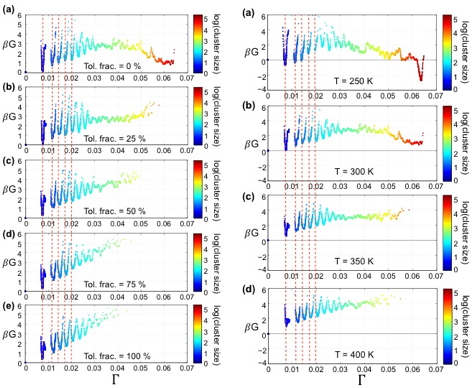
Fig. 2 – Manifold learning discovers a pseudo-1D parameterization of the asphaltene assembly landscape for a prototypical continental asphaltene in a machine-learned order parameter Λ. Each point is a cluster observed in our simulations. The left column shows the effect of n-heptane/toluene solvent composition on the landscape at P=1 bar and T=300 K. The right column shows the effect of temperature at P=1 bar in pure n-heptane. The landscape is modulated in the same manner by elevated temperature and elevated toluene solvent fraction. G is the Gibbs free energy and β=1/kBT is the reciprocal temperature.
Coarse-grained model construction. We have expanded our coarse-grained model construction protocol to archipelago asphaltene architectures (Fig. 3). This demonstrates that our approach can use all-atom simulation data to perform bottom-up parameterization of arbitrary asphaltene architectures. The aggregation behavior of archipelago architectures is well-described by the Yen-Mullins model, but the specific details differ depending on the specifics of the molecular chemistry.
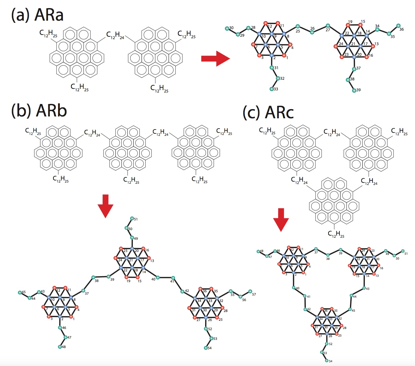
Fig. 3 – Fine- and coarse-grained representations of three prototypical archipelago asphaltene architectures for which we have developed coarse-grained molecular force fields using our bottom-up parameterization protocol. Model ARa is a linear two-core archipelago, ARb and ARc are three-core archipelagos in linear and cyclic topologies.
Archipelago asphaltene folding landscapes. Nonlinear manifold learning applied to the intramolecular configurations of archipelago asphaltenes discovers low-dimensional folding landscapes containing a diversity of metastable configurations with a richness approaching that of protein folding. The folding landscape for model ARb (cf. Fig. 3) is found to exist in three collective machine learned order parameters [Ψ2, Ψ3, Ψ5] that resolve a diversity of metastable folded states respecting the underlying symmetry of the molecule (Fig. 4).
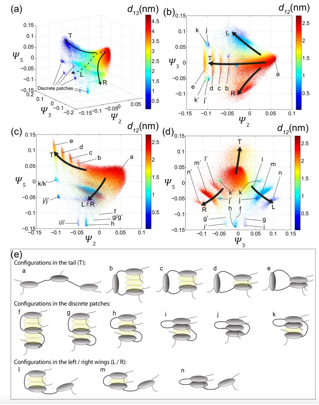
Fig. 4 – Folding landscape for archipelago asphaltene ARb. (a-d) Different projections of the folding landscape into the three machine-learned collective order parameters [Ψ2, Ψ3, Ψ5]. Each point is a particular molecular configuration observed in our simulations, and are colored by dXY denoting the center of mass distance between the Xth and Yth aromatic cores. Particular representative structures are indicated by lower-case letters. (e) Cartoon schematics of the representative molecular configurations.
Professional development. This award has permitted the PI to establish himself within the field of asphaltene self-assembly, and has led to the development of new coarse-grained molecular models and nonlinear manifold learning techniques. The graduate researcher supported by this award, Jiang Wang, has received close mentorship in molecular simulation, statistical thermodynamics, and scientific presentations and publications. Jiang has now published two first author papers under this award, has presented work at the APS March Meeting, and mentored an undergraduate researcher, Mohit Gayatri.










