Reports: UR455140-UR4: Structural Effects on the Primary Isotope Dependence of Secondary Kinetic Isotope Effects in Hydride Transfer Reactions in Solution
 printer friendly
printer friendlyThis proposal examines further consequences of the concept in contemporary H-tunneling models that transferring a heavier isotope requires a shorter donor-acceptor distance (DAD) in the tunneling-ready-state (TRS). The strategy is to investigate how the identity of the transferring “in-flight” primary (1°) H/D isotopes affect the “in-place” secondary (2°) C-H bond vibrations. We determine the 1° isotope effect on the kinetic isotope effect (KIE) at the 2° C-H/C-D positions to examine the 2° C-H bond vibration difference for the transferring of the two isotopes. The hypothesis in the original proposal is that H-tunneling and D-tunneling have tunneling-ready state (TRS) structures which have different DADs, and pronounced 1o isotope effect on 2o KIEs should be observed in the sterically hindered H-tunneling systems (Figure 1). In the past year’s research on this project, we found that in addition to the steric effect that can affect the 1° isotope dependence of the 2° KIEs, the electronic effect such as the H-bonding effect can also affect the 1° isotope dependence of the 2° KIEs at the polarized C-H(D) bond in the TRS. Furthermore, we found that the 2o KIEs at the remote positions could also be sensitive to the 1° isotope.

Figure 1. It is hypothesized that the steric and H-tunneling factors could augment the 1o isotope effect (i.e. the DAD effect) on the 2o KIEs at the various positions. In D-tunneling, the steric effect and H-bonding effect on the 2o H/D vibrations are more pronounced than those in H-tunneling. The oval-shaped area is meant to show the wave-packet of the transferring H/D in the TRSs. The H wave packet is more diffused than the D wave packet.
The system that we mainly study in the past year is the hydride transfer from 10-methylacridine (MAH) and 9,10-dimethylacridine (DMAH) to the 9-aryl xanthylium ions (ArXn+BF4-, Ar = 4-MeOPh and Ph) in acetonitrile (eqn (1)). These reactions are in contrast to the reactions with the tropylium ion (Tr+BF4-) that has much smaller steric requirement, which have been reported before (eqn (2)). In the Tr+ reaction, it was found that the β-2o KIE at the 9-CH3/CD3 position is inverse (<1), and more inverse in the D-transfer process than that in the H-transfer process. In the ArXn+ systems, we found the same trend but greater difference in the 2o KIEs in response to the H vs. D-transfers. We also determined the 1o isotope effect on the 2o KIEs at the remote 10-ε-CH3/CD3 position of both MAH and DMAH for their reactions with Tr+ and ArXn+. We found that the ε-CH3/CD3 2o KIE on MAH is inverse and insensitive to the 1o isotope for reactions with all the carbocations. This is the same for the reaction of DMAH with Tr+ but different from its reaction with ArXn+. In the latter reaction, the ε-CH3/CD3 2o KIE is inverse on H-transfer but normal (>1) on D-transfer, an opposite trend to that found at the β-position.

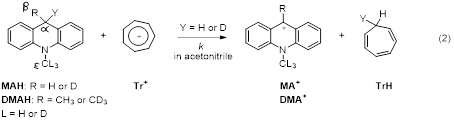
The observation that the 1o isotope dependence of the 2o KIEs at the ε-CH3/CD3 position of DMAH in its reactions with ArXn+ is opposite to the trend at the β-CH3/CD3 position is striking. Our temporary explanation is that the DMAH reaction with the sterically hindered ArXn+ uses a very different TRS conformation from its reaction with MAH. In the TRS, the 9-methyl group of DMAH turns away from the Ar group possibly positioning the polarized C-H bond in the 10-Nδ+-CH3 group to face the Ar group. This may form a H-bonding interaction, and the interaction is pronounced in the D-tunneling that has a shorter DAD. The pronounced H-bonding in D-tunneling results in an inflated 2o KIE. Further studies to confirm the explanation include the study of the 1o isotope dependence of the ε-CH3/CD3 2o KIEs with different hydride acceptors.
Impact of the Research
This is a brand-new physical-organic-chemistry research direction that studies the isotopologues─DAD─TRS structures relationship for the H-transfer reactions. The significance of this project has multiple facets. First, the confirmed isotopically different DAD concept could not only support the activated H-tunneling model, but also provide a foundation for the development of other necessary models for H-transfer Chemistry. Second, the “DAD concept” will change our understanding of the nature of H-tunneling and the origin of primary (1°) KIEs. Third, the “DAD concept” will also help understand the 1° isotope effect on secondary (2°) KIEs that has been previously widely used to provide insights into the H-tunneling mechanism in enzymes. Fourth, the 1° isotope effect, i.e. the DAD effect, on the 2° KIEs at various positions gives the DAD─TRS structure relationship, which can assist the understanding of the DAD sampling coordinate of the H-tunneling reactions. This much-needed study is expected to change many traditional views about KIEs and the H-transfer reactions, and the similar study in enzymes would help understand the H-transfer chemistry in biological systems. The published and preliminary results will help the PI(s) prepare the NSF or NIH grant proposals that will expand the study to more hydride transfer reactions and furthermore to proton and hydrogen-atom transfer reactions.
Undergraduate and MS degree graduate students involved in this project learned basic organic synthesis, kinetic procedures, mechanistic analysis techniques, computational chemistry and the use of modern analytical instrumentation. The knowledge and techniques will make students competitive in their future careers. With these research experiences students are prepared to enter professional schools and Ph.D. programs, to become teachers, and problem-solvers for chemical companies. For example, a MS student partially supported by the grant is now in the UC Davis for Ph.D. study, and an undergraduate student under the support currently studies pharmaceutical sciences in the SIUE pharmacy school.










