Reports: ND756280-ND7: Stimuli Responsive Assemblies of Polymer-Based Colloids
 printer friendly
printer friendlyThe initial proposal focused on an innovative stimuli-responsive-based method to study tunable colloidal self-assembly. Over the past 15 months, one graduate student, Mingzhu Liu, and one postdoctoral fellow, Dr. Beth Elacqua, have realized this goal (Dr. Elacqua started her independent career as an Assistant Professor at Penn State last months. Her training under this grant was essential to secure her current position). We have created light stimuli responsive particles that can be assembled. These results have been published in ACS Macro Letters this month. Below, we introduce these results and discuss details.
The preparation of polymeric colloidal particles has received considerable attention in the last decade, owing to desirable applications in catalysis and drug delivery, and as functional materials for plasmonics and photonics. The majority of these applications require monodisperse particles that are uniform in not only shape and size, but also composition and surface properties. Spherical colloidal particles have emerged as prototypical models since they feature highly tunable, yet inexpensive platforms with the ability to control size, shape, and bonding interactions, while giving rise to a variety of materials that exhibit both long and short range order.
Employing molecular recognition is an advantageous platform for the synthesis of colloidal architectures. For one, the directional strategy is versatile, as multiple complementary motifs, differing in connectivity and association strengths, are available. Supramolecular interactions are also modular and can be easily fine-tuned and regulated with a stimulus. In addition, their reversible nature can lead to adaptive and responsive materials. Whereas supramolecular engineering is utilized routinely in the solid state and polymer chemistry, the exploitation of molecular recognition to engineer larger colloidal architectures is still in its infancy. We have demonstrated the ability to endow bonding specificity and directionality into colloidal particles. Central to our approach is the ability to fabricate molecular geometry-mimicking patchy particles to integrate desired molecular recognition elements in a site-specific manner, while installing directional interactions to manipulate supramolecular assembly. In all cases, the assembled architectures were dictated by the patch geometry.
The research carried out under the PRF grant introduces a directional, controllable and versatile method to assemble colloids based upon the combination of host-guest complexation and synthetic polymer chemistry. Common hosts based upon supramolecular containers such as cucurbit[n]urils (CB[n]) have been assembled with a variety of electron poor (e.g., adamantylammonium and viologen moieties) and/or electron rich (e.g., azobenzene, naphthol) guest molecules. While CB[8] is well-established to fabricate self-assembled polymers and dendrimers, the exploitation of CB[8] to controllably direct the self-assembly of colloids had never been explored. Our research closes this gap.
Our research design is based on a polymer platform to install fluorescently-labeled polymers that are capable of host-guest complexation and a directed disassembly in the presence of a noninvasive stimulus (i.e., light). The use of polymer chemistry is key to the design, as all of the requirements for directional (and observable) molecular recognition are addressed using synthesized monomers, initiators, and terminators. In our design, ring-opening metathesis polymerization (ROMP) is utilized owing to the ability to introduce a variety of functionalized norbornenes and initiators that facilitate directional self-assembly, while maintaining a controlled polymerization. By exploiting ROMP, the density of molecular recognition elements can be altered with target block length, while functional initiators and terminators provide versatility to the process.
We synthesized telechelic polymers via a functionalized Grubbs initiator leading to block copolymers comprising viologen (Vio) and azobenzene (Azo) pendant side-chains (Figure 1). The block copolymers also contain spacer blocks, a hydroxylated terminus, and aldehyde motifs. The aldehydes are functional handles that can be conjugated to a cyanine (Cy3 or Cy5) dye via post-polymerization reactions to afford a terminal moiety that confirms self-assembly specificity in the presence of CB[8]. The alcohol groups installed through the initiator covalently attach the polymers to carboxylic acid-functionalized patches of anisotropic particles in a site-specific manner (Figures 1 and 2).
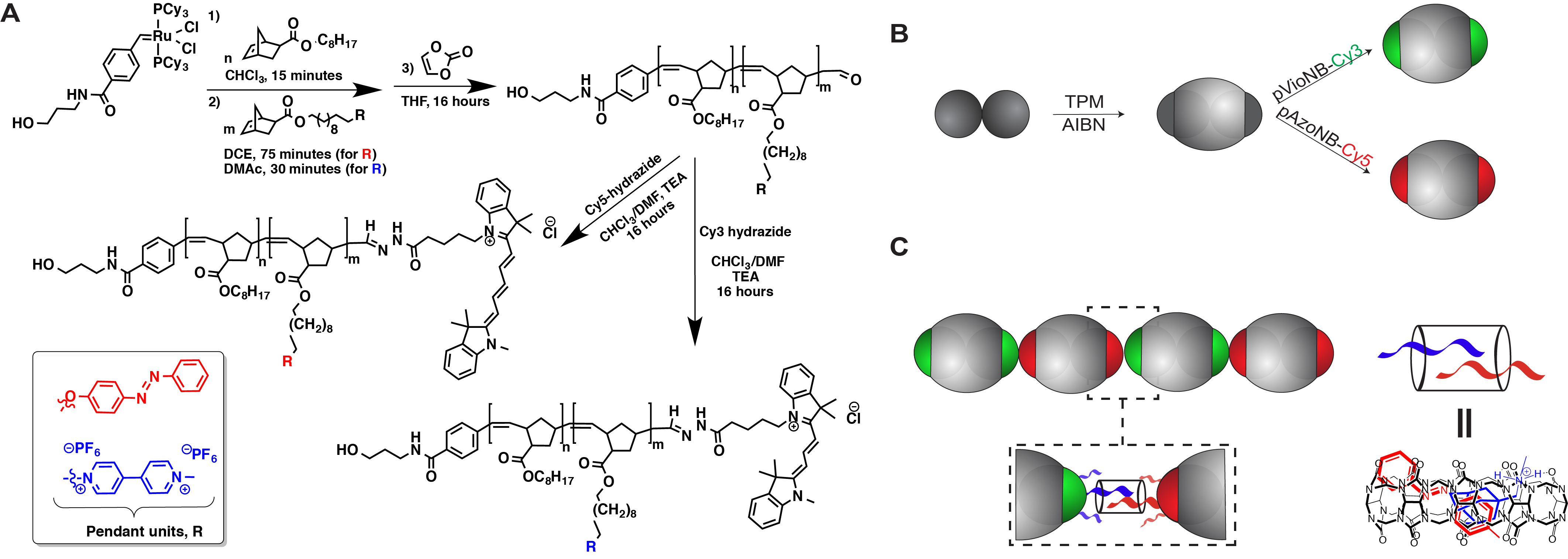
Figure 1. Host-guest strategy featuring both synthetic polymer chemistry and colloidal assembly: (A) engineering of heterotelechelic side-chain-functionalized block copolymers through ROMP; (B) fabrication of patchy particles from carboxylated poly(styrene) colloidal clusters through encapsulation and polymerization with 3-(trimethoxysilyl)propyl methacrylate (TPM), followed by polymer conjugation; and (C) target host-guest driven assembly mediated by cucurbit[8]uril.
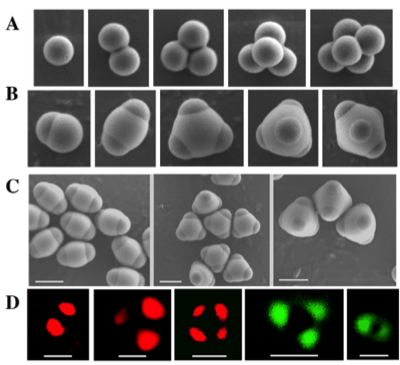
Figure 2. Scanning electron micrographs of (A) carboxylated poly(styrene) clusters, (B) TPM-encapsulated patchy particles, and (C) purified batches of dimers, trimers, and tetramers, and (D) confocal micrographs of individual (red) pAzoNB-Cy5 and (green) pVioNB-Cy3 functionalized patchy particles (scale bars = 1 μm).
Host-guest complexation of pendant Vio and Azo moieties in the presence of CB[8] induce self-assembly and afford colloidal clusters and chains (Figure 3). The Vio and Azo recognition units are utilized as receptors owing to strong associations with CB[8]. Our choice of Vio and Azo functionalities is founded upon the ability to facilely integrate both groups within a norbornene backbone, while fabricating a switchable anisotropic particle assembly that can be controlled with light.
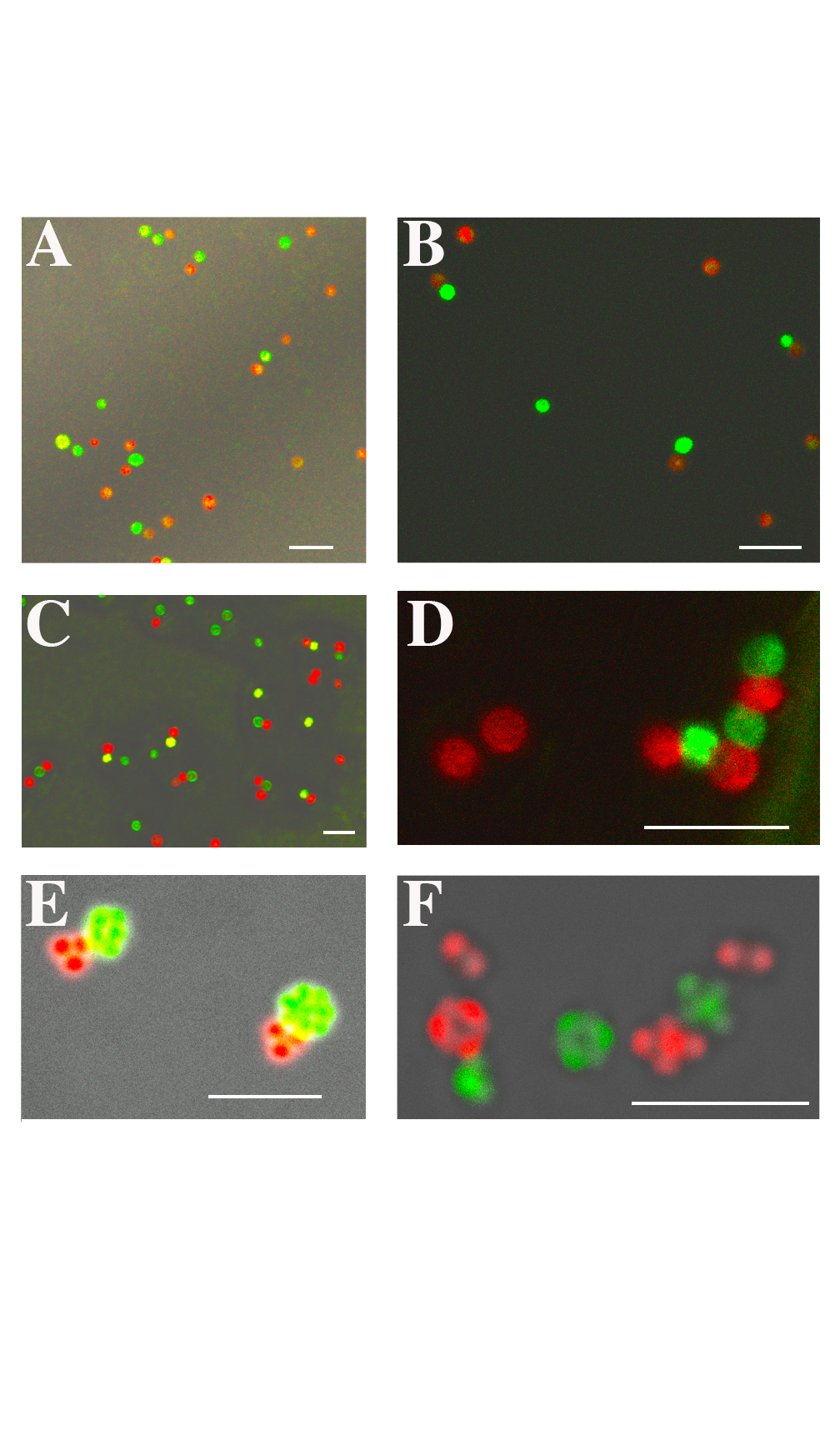
Figure 3. (left) Bright-field and confocal overlay and (right) confocal images of self-assembled particles: (A, B) monopatch, (C, D) dipatch, and (E, F) mixed patch assemblies depicting clear patch-patch interactions (scale bars = 5 μm).
The resultant complexes undergo reversible disassembly/self-assembly in the presence/absence of UV light (Figures 3 and 4). Owing to a difference in time scale of the CB[8] related assembly/disassembly/reassembly process with Azo motifs, we imagine that the integration of these host-guest molecular recognition elements into higher valency particles has the potential to provide a facile and noninvasive method to reconfigure 3D particle assemblies.
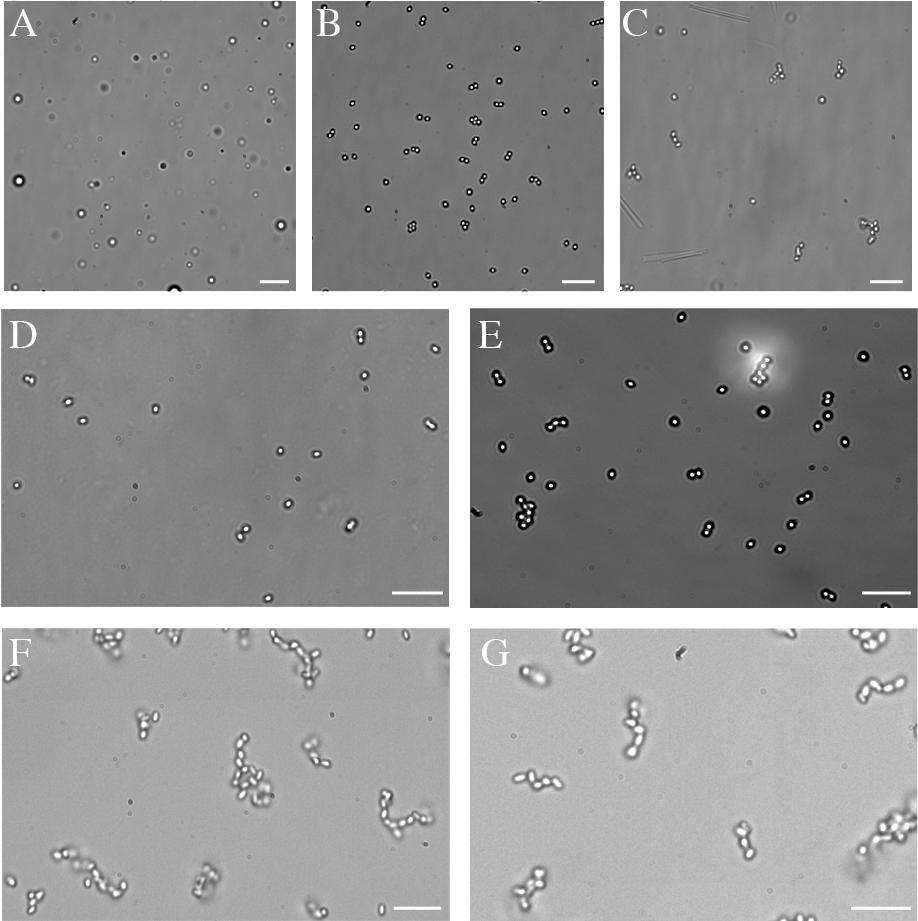
Figure 4. Bright-field images illustrating reversible nature of host-guest interactions: (A) dimer particles prior to assembly; (B) clustering effects seen after 15 minutes mixing with CB[8]; (C) assembled chains forming after two hours; and (D) particle mixture after eight hours of UV irradiation; and (E) reassembled particles after eight hours of visible light irradiation.
In summary, this report describes the use of host-guest chemistry to drive the directional self-assembly of functionalized colloidal particles into open structures. The resultant complexes undergo reversible disassembly/self-assembly in the presence/absence of UV light.










