Reports: UR355304-UR3: Stoichiometric and Catalytic Nitrene-Group-Transfer Reactions from Late-Metal Silylamides
 printer friendly
printer friendlyNOTE: As I communicated to Nancy Jensen in May 2016, Carleton has begun a science facility project during the past year that will ultimately give my lab significant amounts of new space but has temporarily displaced us. As a result of my old labs being demolished this year, I was unable to take students to work on my PRF project during most of the academic year and all of summer 2017. This is reflected in the budget for the past year and also in the smaller number of results reported herein, which were obtained during the time my lab was running prior to March 2017. We have now been mostly moved into a temporary lab and plan to resume our PRF project beginning in January 2018.
In this project, my research group is investigating the possibility that late-metal silylamides, particularly those of base metals, can serve as precursors to reactivity imido or related species that will participate in nitrene-group transfer. One goal of this work is to provide insights into how oxidative deprotection can be used as a strategy for unmasking reactive nitrogen groups in the presence of an appropriate metal to promote the reaction. A proposed catalytic cycle is shown in Figure 1, and we are particularly excited about the possibilities afforded by one-electron oxidation/deprotection steps to facilitate this reactivity at base metals, potentially in an electrocatalytic fashion.
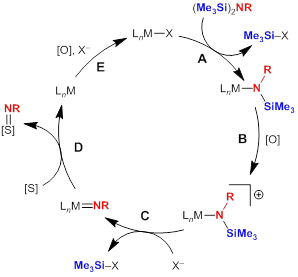
Figure 1. Possible catalytic cycle utilizing late-metal silylamides as protected nitrene-group sources
Studies of (dtbpe)Ni Silylamides and their Redox Properties
We have initially targeted nickel silylamides supported by the 1,2-bis(di-tert-butylphosphino)ethane (dtbpe) ligand for exploration of steps B and C in the cycle from Figure 1. The silylamide complexes were prepared in high yield from the previously reported bis-μ-chloride dimer (Figure 2), and crystal structures have been obtained for two amides.
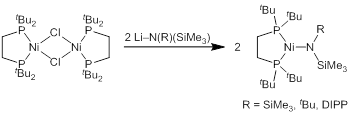
Figure 2. Synthesis and structures of (dtbpe)Ni silylamides
All three complexes show an electrochemically reversible oxidation to Ni(II) (step B in the proposed cycle). Furthermore, we were encouraged to see that introduction of a silyl group on the amide (versus hydrogen) led a more oxidatively stable complex without the additional irreversible feature exhibited by Hillhouse’s hydrogen-substituted amide (Figure 3).
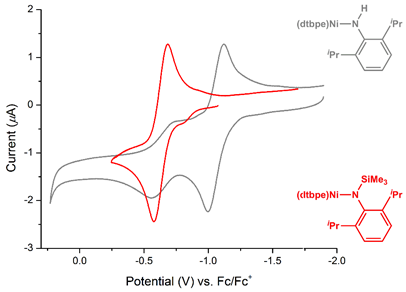
Figure 3. Voltammograms of (dtbpe)Ni amides with varying substitution
Our efforts during the past year have been focused on chemically oxidizing the silylamide in order to understand the possibility of silyl-group removal from the oxidized species (step C in Figure 1). Oxidation with ferrocenium and silver(I) oxidants leads to the same major product, which we formulate as the cationic amide complex, tentatively supported by regeneration of the neutral amide upon introduction of cobaltocene to re-reduce the cationic product.
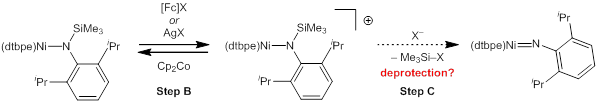
Figure 4. Oxidation of (dtbpe)Ni–N(TMS)(DIPP) and proposed deprotection
During attempts to crystallize the oxidized product as a BF4– salt, we observed an unexpected decomposition to a Ni(II) fluoride complex (Figure 5).
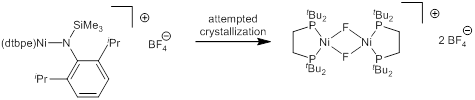
Figure 5. Decomposition of oxidized nickel silylamide in the presence of BF4–
This unexpected decomposition may be promoted by interaction of fluorides from BF4– with the silyl group, which would suggest that Step C could be feasible. However, the mechanism at this point is unclear. We have prepared ferrocenium oxidants with unreactive and highly soluble tetraarylborate counterions and found that oxidation proceeds similarly but affords much more stable products. Over the next year, we will explore the reactivity of these soluble Ni(II) cationic amides, with an eye toward preparing reactive Ni(II) imido complexes from them.










