Reports: DNI355989-DNI3: A Structural Diphosphine Model to Predict Metal-Phosphorus Covalency Variations in Transition Metal Complexes
 printer friendly
printer friendlyThe ACS Petroleum Research Fund supported P K-edge X-ray absorption spectroscopy (XAS) experiments aimed at determining how changes in diphosphine ligand structure affect covalency in metal-phosphorus (M-P) bonds. Diphosphines are very important ligands in homogeneous transition metal catalysis, and our goal is to develop a general structural diphosphine model to predict variations in M-P covalency. We postulate that M-P covalency, which is known to govern certain aspects of chemical reactivity, will yield actionable structure/reactivity comparisons that can be used to predict catalyst function.
We completed the following experiments at the University of Iowa and the Stanford Synchrotron Radiation Lightsource (SSRL) in Menlo Park, CA in 2016-2017. The experiments were aimed at addressing three objectives of our proposed work.
1. We collected P and Cl K-edge XAS data on square planar [Cy2P(CH2)nPCy2]PdCl2 complexes (n = 1 – 3) for comparison to [Ph2P(CH2)nPPh2]PdCl2. The objective was to determine how changing the phosphorus substituents from phenyl to alkyl affected trends in M-P covalency.
2. We collected P and Cl K-edge XAS data on [Ph2P(CH2)nPPh2]TiCl4 complexes (n = 1 – 3). The objective was to determine if established structure-induced trends in M-P covalency could be observed for complexes containing metals other than Ni and Pd.
3. We collected P K-edge XAS data on [Ph2P(CH2)nPPh2]PdCl2 solutions (n = 1 – 3) using an XAS solution flow cell designed in collaboration with Los Alamos National Laboratory. The objective was to determine if solid-state P K-edge XAS results are representative of XAS data collected in solution where homogeneous catalysts operate.
Objective 1. In preliminary work that was published after submission of the ACS-PRF proposal, we showed that changes in diphosphine bite angle (P-M-P; β) do not track with anticipated changes in M-P covalency in a series of Pd diphosphine complexes (Figure 1). We discovered a 10% increase in M-P covalency for [Ph2P(CH2)2PPh2]PdCl2 over the other three compounds in the study (Figure 2). For these and the Pd compounds described below, M-P covalency correlates to the first peak intensity in the P K-edge spectra.
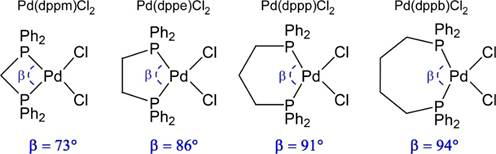
Figure 1. P-M-P angle (β) comparisons in [Ph2P(CH2)nPPh2]PdCl2 complexes (n = 1 – 4).
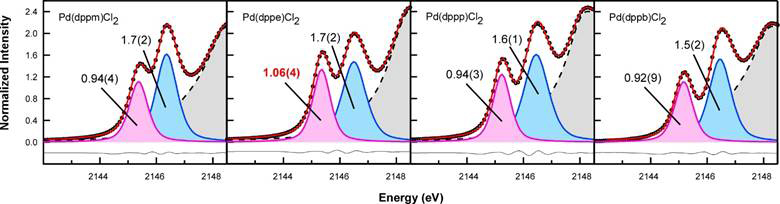
Figure 2. Curve fit models of P K-edge XAS spectra collected for [Ph2P(CH2)nPPh2]PdCl2 (n = 1 – 4).
The P K-edge XAS spectra of the cyclohexyl-substituted diphosphine complexes [Cy2P(CH2)nPCy2]PdCl2 showed the same trend in M-P covalency for [Ph2P(CH2)nPPh2]PdCl2 complexes in Figure 1. Peak intensities were identical within error as the number of CH2 linkers in the diphosphine backbone were increased from 1 to 3 (Figure 3). This indicated that changing the identity of the phosphorus substituent from phenyl to cyclohexyl has no effect on M-P covalency within the error of the measurement. Instead variations in the M-P covalency correlate to changes in the diphosphine backbone and can be modeled using only the P-M-P and M-P-C angles. We call this the “phosphine twist model”.

Figure 3. Curve fit models of P K-edge XAS spectra collected for [Cy2P(CH2)nPCy2]PdCl2 (n = 1 – 3).
Objective 2. We collected P K-edge XAS data on octahedral [Ph2P(CH2)nPPh2]TiCl4 complexes (n = 1 – 3) containing the same diphosphines shown in Figure 1. Our aim was to determine if similar trends in M-P covalency could be observed with different metals as the diphosphine backbone was modified. [Ph2P(CH2)PPh2]TiCl4 was not known prior to our work and [Ph2P(CH2)3PPh2]TiCl4 was not structurally characterized. Hence, we prepared and determined the structures of all three compounds before proceeding with XAS experiments (Figure 4).
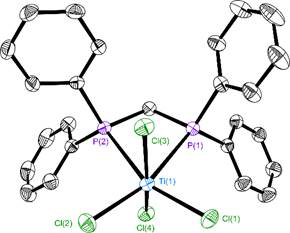
Figure 4. Molecular structure of [Ph2P(CH2)PPh2]TiCl4 with ellipsoids drawn at the 35% level. Hydrogen atoms were omitted from the figure.
Unlike our studies with Pd, P K-edge XAS data collected on the Ti complexes revealed that transitions associated with M-P covalency are not resolved from transitions associated with phosphorus-carbon bonds on the ligand (Figure 5). Unfortunately, this has so far prevented us from quantifying the intensity of the transitions, but time-dependent density functional theory calculations suggest that M-P covalency predictions made using the phosphine twist model hold. Based on our Pd studies and preliminary DFT calculations, we postulate that changing the phosphorus substituents from phenyl to alkyl will allow us to resolve the desired pre-edge P K-edge features the Ti complexes. These efforts are in progress.
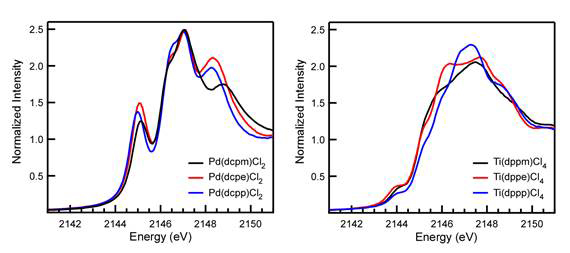
Figure 5. Stack plot comparisons of P K-edge XAS data for [Cy2P(CH2)nPCy2]PdCl2 (left) and [Ph2P(CH2)nPPh2]TiCl4 (right).
Objective 3. P K-edge XAS data is typically collected on solid samples because solution data collection is hampered by photon-induced sample decomposition and evaporation of the solvent. We prepared a solution flow cell to circumvent these issues (Figure 6), and used it to successfully collect P K-edge data on dichloromethane solutions of [Ph2P(CH2)nPPh2]PdCl2 (n = 1 – 3) for comparison to their solid state data. The flow cell removes decomposition products as they are generated and attenuates solvent heating and evaporation.

Figure 6. Flow cell designed for solution P K-edge XAS studies.
The P K-edge XAS data revealed that the 10% increase in M-P covalency for [Ph2P(CH2)2PPh2]PdCl2 in the solid-state data disappeared in solution. There is no statistically significant difference in the first peak intensity for all three compounds, which suggests a significant change in M-P covalency for [Ph2P(CH2)2PPh2]PdCl2 in solution. We are currently investigating why this change occurs.
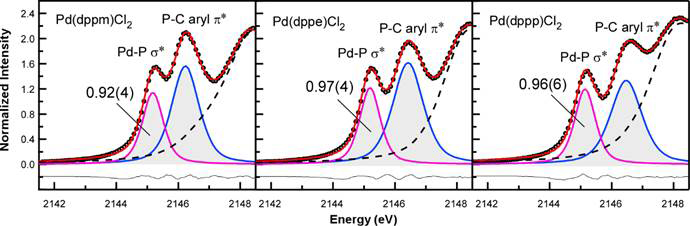
Figure 7. Curve fit models of solution P K-edge XAS spectra collected for [Ph2P(CH2)nPPh2]PdCl2 in dichloromethane.
Impact of research. Funding from the ACS PRF provided critical early career support to build a dedicated XAS program aimed at modeling M-P bonding and electronic structure in metal complexes. In addition to achieving the outlined objectives described here, PRF support has provided institutional knowledge, student training, and travel support needed to pursue parallel P K-edge XAS studies. New diphosphine catalysts prepared in the Daly Group, as well as complexes containing other phosphorus ligands have been explored. The latter work garnered important early career recognition. We were awarded the journal cover for the Dalton Transactions “New Talent: Americas” issue for PRF-supported studies of Rh PNP pincer complexes. Three manuscripts describing work in this report are in various stages of the publication process (one submitted, two in preparation).










