Reports: ND655026-ND6: Understanding the Relationship between Fluid Molecular Structure and Pressure-Viscosity Behavior
 printer friendly
printer friendlyThis project is focused on understanding the connection between the viscous properties of lubricants and lubricant additives and the structure and chemistry of their constituent molecules. During the second year of the project, we focused on the important role of viscosity modifiers (VM)s, which are long chain polymers added to lubricant formulations to improve their viscous properties during operation. The goals of our research efforts this year were to (a) understand how the structure of a polymer affects its ability to increase viscosity at high temperatures, and (b) explore the fundamental mechanisms by which polymeric VMs increase viscosity.
To understand structure-function relationships, we studied styrene-butadiene (see Figure 1a) in three different configurations, alternating, random, and block (see inset of Figure 1b), all having approximately the same molecular weight of 3900 g/mol. The viscosity-temperature response of the polymers in a dodecane solvent was characterized using molecular dynamics simulation. First, viscosity was obtained from simulations at a range of high shear rates and the resultant data was fit to the Carreau equation to calculate the Newtonian viscosity (see Figure 1b). The temperature-viscosity behavior of each fluid was quantified by its proportional viscosity index, which indicated that the block configuration exhibited the best viscosity-temperature behavior, i.e. the least decrease of viscosity with increasing temperature. To understand this result, the radius of gyration and the intramolecular interactions of each polymer model were characterized as functions of temperature. Trends in coil size and contact pairs were consistent with those observed in the viscosity index. Of the three configurations studied, the block structure exhibited the least change in viscosity with temperature. This configuration was also observed to form small and tight coils with many intramolecular interactions at low temperatures, and then expand as the temperature was increased (see Figure 1c). This observation suggests that structures that can attain very small conformations at the lower temperatures and expand as temperature is increased will function as ideal VMs. In general, these results indicate that VM behavior is strongly correlated to polymer structure because the structure determines the polymer's response to temperature. Further, the method proposed in this study can be applied to other chemistries to better understand the structure-property relationships that are key to VM performance. Successful implementation may lead to the development of a molecular tuning tool which could redefine our understanding of fluid properties and facilitate the design of lubricants with optimal performance and functionality.
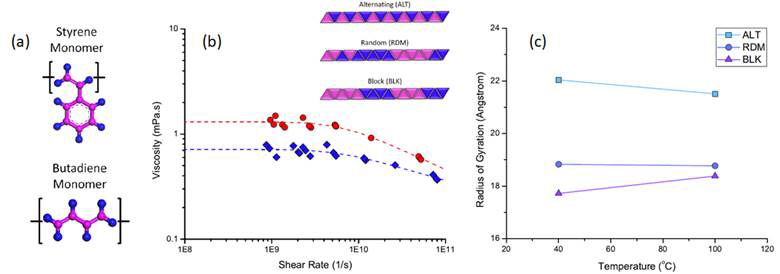
Figure 1: (a) Illustrations of the styrene and butadiene monomers that comprise all three polymer models. (b) Viscosity of solution containing dodecane and the random styrene-butadiene structure at 40 and 100C, where the simulation data (symbols) are fit to the Carreau equation (dashed line) to calculate Newtonian viscosity. The inset is a schematic illustration of the three polymer structures modeled. (c) Mean radius of gyration of each polymer at the two temperatures.
To explore the fundamental mechanisms by which polymeric VMs increase viscosity, we used atomistic models of polyisobutylene (PIB) polymer in polyalphaolefin (PAO) base oil. The simulations of 1 and 2 PIB molecules in PAO at 10 wt.% (see Figure 2a) were used to obtained Newtonian viscosity as described in the previous paragraph (see inset to Figure 2c). The accuracy of the models was validated by comparison to experimental measurements and then the simulations were used to explore the mechanisms by which the PIB increased the viscosity of the solution. First, the role of coil expansion was characterized as the polymer's radius of gyration. No shift in the distribution towards larger radii of gyration with temperature was observed, which indicates that the coil size mechanism does not contribute to the effect of PIB on solution viscosity. Next, the effect of association between PIB molecules was quantified as the number of atoms “in contact” (see inset to Figure 2b). There was more polymer-polymer contact at 100C than 40C, which may suggest that association is a contributing factor. This hypothesis was then tested by creating a fictitious model system in which the two PIB molecules interacted with each other only via repulsive forces. The new model successfully minimized the number of contact atoms (Figure 2b), but did not affect the solution viscosity (Figure 2c), which indicates that the association mechanism does not play a major role in PIB function as well. Lastly, we studied the effect of the PIB on the alignment of the PAO using the length of the PAO in the shear direction as a simple approximation of molecule orientation. A comparison of the lengths with and without the PIB present revealed more PAO molecules aligned with the flow when the PIB was present. Furthermore, less-aligned PAO molecules were more likely to be found closer to the PIB than further away. Both of these analyses suggest that the PIB may be increasing viscosity indirectly through its effect on nearby PAO.
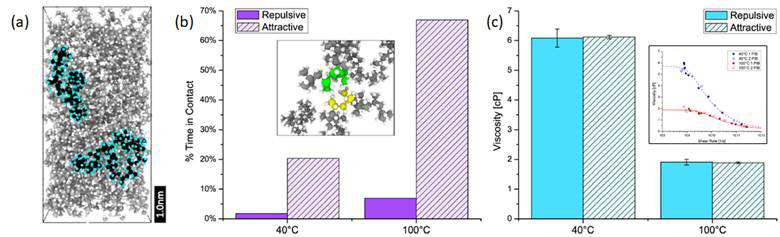
Figure 2: (a) Snapshot of the simulation with 2 PIB molecules in PAO. (b) Percent of time the two polymers are “in contact”; inset shows how contact between atoms is identified. (c) Viscosity of the solution with and without attractive interactions between the two polymers; inset shows viscosity data from simulation (symbols) fit to the Carreau equation (dashed lines).
References:
U.S. Ramasamy, M. Len and A. Martini, Correlating Molecular Structure to the Performance of Linear Styrene-Butadiene Viscosity Modifiers, Tribol. Lett. Accepted
M. Len, U.S. Ramasamy, S. Lichter and A. Martini, Thickening Mechanisms of Polyisobutylene in Polyalphaolefin, Tribol. Lett. Under Review










