Reports: DNI156575-DNI1: A Reverse Polarity Approach to Preparing Chiral Amines through Stereoselective C-C Bond Assembly
 printer friendly
printer friendlyI. Introduction
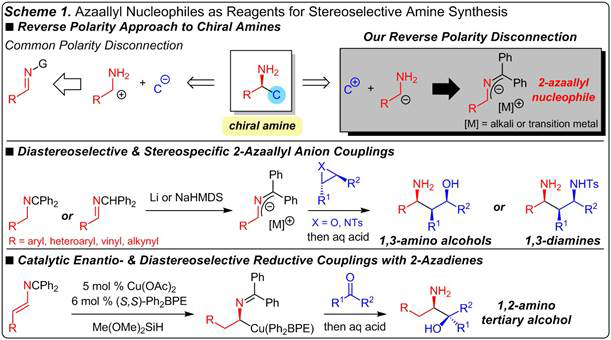
Research during the first year of this award has focused on stereoselective umpolung approaches to generating chiral amines that are challenging to prepare through the normal polarity paradigm (Scheme 1). We have pursued two distinct but related tracks for achieving this aim, the second of which is a new direction that has evolved from the first. 1) On the first path, we have investigated reactions of 2-azaallyl anions, nucleophilic alpha-amino anions that are derived from electrophilic imines. In the original grant proposal, we described two types of reactions that would be developed with azaallyl anions: Pd-catalyzed enantioselective allylic substitution and diastereoselective and stereospecific additions to epoxide and aziridine electrophiles. Although we failed to improve upon initial results in the catalytic allylic substation, we successfully developed two methodologies related to couplings with epoxides and aziridines and have published two research articles describing that work. 2) Our second research direction stemmed from limitations encountered in azaallyl anion chemistry, namely the need for strong base, the inability to promote reactions with alkyl-substituted nucleophiles, and the requirement of substrate control over regio- and stereochemical outcomes. We have therefore introduced 2-azadienes as new reagents for catalytically enantioselectively preparing chiral amines. Migratory insertion of the azadienes to form azaallyl transition metals represents umpolung of an enamine and constitutes a new mode for generating alpha-aminoalkyl transition metals. The azaallyl transition metals may be combined diastereo- and enantioselectively with various electrophiles.
II. Diastereoselective and stereospecific additions of 2-azaallyl anions to epoxides and aziridines
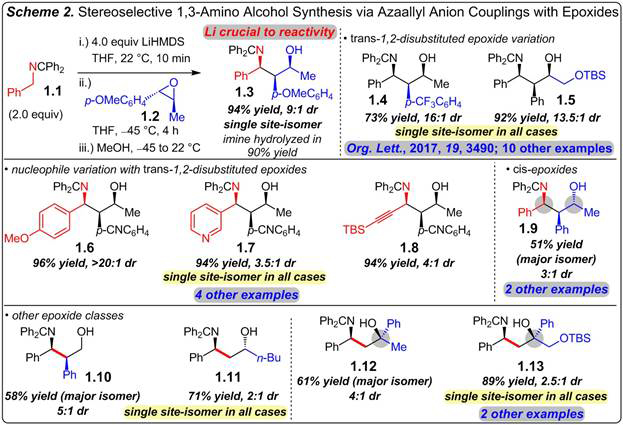
We have developed a method that couples azaallyl anions and epoxides with excellent levels of stereocontrol and site-selectivity to furnish 1,3-amino alcohols with 2–3 stereogenic centers in good yield (Scheme 2). Ketimine 1.1 is deprotonated with LiHMDS and added to enantiomerically enriched trans-epoxide 1.2; MeOH quench then affords 1,3-amino alcohol 1.3 in 94% yield as a single site-isomer in 9:1 dr. The use of the ketimine tautomer of the azaallyl anion precursor and a Li base proved crucial for obtaining reaction. The aldimine tautomer cannot be deprotonated efficiently with LiHMDS and Na and K anions lead to <2% conv under otherwise identical conditions, a significant difference from tosylaziridines (see Scheme 3). A fluorenyl group in place of benzhydryl leads to reaction only at the undesired anion position. Several trans-aryl/alkyl-1,2-disubstituted epoxides are competent electrophiles, leading to coupling at the least-hindered nucleophile carbon and the epoxide’s benzylic position (1.4–1.5). Several nucleophilic partners participate to afford 1,3-amino alcohols with a variety of substituents at the N-bearing center (1.6–1.8). cis-Epoxides react with azaallyl anions, albeit more slowly to deliver amino alcohols that are epimeric at the carbinol center (1.9). Although diastereoselectivity is modest, in part due to the elevated temperature, the major isomer may be selectively isolated (51% yield) after purification, a hallmark of this method. Primary alcohol 1.10 is formed exclusively via benzylic ring-opening with the major isomer isolated in 58% yield; comparatively, n-hexene oxide leads to 1.11, formed from terminal carbon attack (bond formations in red). 2,2-Disubstituted epoxides undergo ring-opening at the terminal position, in contrast to the analogous tosylaziridines (see Scheme 3), to deliver tertiary alcohols 1.12 and 1.13. Reactions are efficient and the major stereoisomer may be selectively isolated. Amino alcohols that are not easily prepared via Mannich reaction or C–H amination may be accessed via this method.
We have also established that azaallyl anions may diastereoselectively and stereospecifically add to several aryl-substituted classes of chiral aziridines (Scheme 3). Benzylketimine 1.1 or benzhydrylaldimine 1.14 may be deprotonated efficiently with NaHMDS and added to phenyl-substituted tosylaziridine 1.15 to deliver differentially-protected 1,3-diamine 1.16 as a single site-isomer in 8:1 dr and 90–95% yield. Reaction takes place exclusively between the less hindered position of the anion and the aziridine’s benzylic carbon, occurring with complete inversion of stereochemistry. Hydrolysis of 1.16 under mild conditions furnishes free primary amine 1.17, isolated as one diastereomer after a single recrystallization. A Na counterion proved optimal with terminal aziridines; reactions with Li or K are either less efficient or diastereoselective.
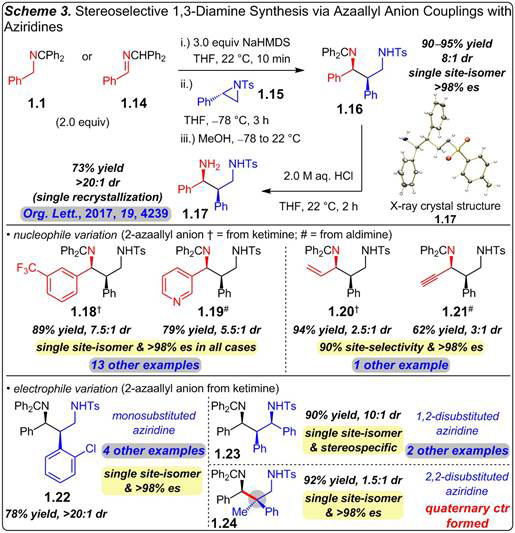
Our studies show several aryl- or heteroaryl-substituted azaallyl anions may be added to aziridine 1.15 in 69–93% yield with 5.5–9.5:1 dr (e.g., 1.18–1.19); all are obtained as a single site-isomer and enantiomer. Vinyl- and alkynyl-substituted anions may also be utilized to obtain allylic/propargylic amines 1.20–1.21 stereoselectively (2.5–3 dr), stereospecifically (>98% es), and site-selectively (80–90%). Variations in the aziridine’s aryl group lead to diamines in up to >20:1 dr (1.22). 1,2-Disubstituted aziridines participate in azaallyl anion couplings to deliver 1,3-diamines bearing three contiguous stereogenic centers (1.23). Azaallyl anion reaction with a 2,2-disubstituted aziridine still results in attack exclusively at the benzylic center to afford 1.24 (bond formation in red), which contains a quaternary carbon, in 92% yield. Each diastereomer is formed with >98% es from the enantioenriched aziridine.
III. Catalytic enantio- and diastereoselective reductive couplings of 2-azadienes and ketones
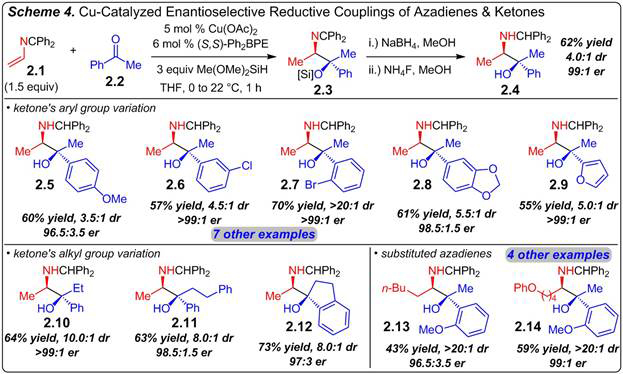
We have established a method for the reductive coupling of 2-azadienes with ketones and imines (Scheme 4). Aryl/alkyl ketones, such as acetophenone 2.2 undergo coupling with azadiene 2.1 and dimethoxymethylsilane within 1 h in the presence of a chiral Cu catalyst prepared from Ph2BPE to form imine 2.3. For ease of handling and analysis, the product’s imine is reduced to the secondary benzhydrylamine and the hydroxyl group unmasked from the silyl ether to generate amino alcohols: compound 2.4 is thus formed in 4:1 dr with the major diastereomer isolated in 62% yield and 99:1 er (both diastereomers are formed with identical enantioselectivity). Variation of the ketone’s aryl group (2.5–2.9) or alkyl group (2.10–2.11) are tolerated; cyclic ketones participate efficiently in the reaction (2.12). Diastereoselectivity is good and is improved with ortho-substituted aromatic rings or alkyl groups larger than methyl (3.5 to >20:1 dr, 45–87% yield). 4-Substituted azadienes generate products with alpha-alkyl groups other than methyl (2.13–2.14). Enantioselectivity is excellent in all cases (>95:5 er). Future work will focus on other enantioselective couplings of azadienes.