Reports: DNI1057003-DNI10: Toward Chemical Accuracy for the Description of the Catalytic Desulfurization Process
 printer friendly
printer friendlyThe goal of the project
The aim of this project is to provide a deep quantitative understanding of weak interactions in the catalytic desulfurization process. The catalytic reactions begin with the adsorption of the sulfur containing compound, and then the molecule undergoes a series of reactions which leads to the C-S bond scission.
Surfaces of metal oxides, and in particular transition metal oxides, play an extremely important role in heterogeneous catalysis, either as active catalysts or as support for nanostructured metallic catalysts. The adsorption of organic molecules on metal surfaces requires a proper modeling of weak nonlocal van der Waals (vdW) correlation.
In the first year of the grant, our focus has been on establishing benchmark methods. Our work on the methodology and tests with the Random Phase Approximation (RPA) can be regarded as an alternative to the originally proposed MBD-vdW method. RPA-based methods have become recently available and applicable with the GPAW (PAW) code. On the other hand we have become aware of some numerical problems related to the MBD-vdW approximation in certain electronic structure codes. RPA has the same physics relevant to catalytic adsorptions as MBD-vdW. Although our recent work with RPA and beyond-RPA methods is a minor deviation from the originally proposed method, it still can play the same role as MBD-vdW for the estimation of a benchmark vdW approximation for surface energies and adsorption processes.
1. Benchmark surface energies within Renormalized Random Phase Approximation: All of the interactions related to the adsorption of an organic compound are occurring on metal or semiconducting surfaces. When the adsorption energy is computed, one has to rely on the clean surface energy as a contribution to the adsorption energy. Although surface energies have been extensively obtained with semilocal density functionals, the quantitative accuracy of these approximations is questionable. There is a high need for a quality control with reasonable computational cost/accuracy ratio. In my research group we have applied the RPA within the density functional context. Due to the efficient implementations and increasing computer power, RPA has become an applicable method in materials science. RPA has the fully nonlocal long-range correlation relevant for capturing vdW interactions. Its correlation energy is however too deep at the short range. This deficiency can be improved by a kernel correction through the Dyson equation for the screening. Kernels can be derived from many-body approximations or can be modeled by satisfying exact constraints. In our work we resort to the latter method. Solving the Dyson equation with kernel can however create instability. To circumvent this problem we can resort to RPA renormalization. Within this approach we make a linear approximation in powers of the kernel. The renormalized RPA (RPAr1) avoids the divergence
for small gap systems and converges fast.
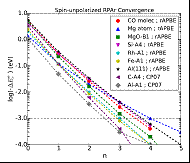
Figure 1: The convergence behavior of the Renormalized RPA approximation for various systems.
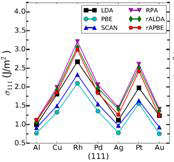
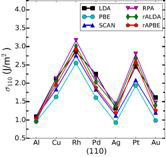
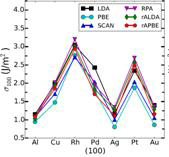
Figure 2: Surface energies for the three lowest energy crystal faces with various density functional approximations. RPA refers to the infinite-order RPA while rALDA and rAPBE are the model kernels chosen within the RPAr1 approach.
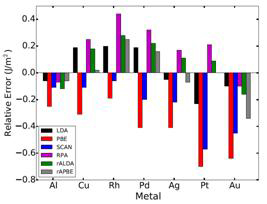
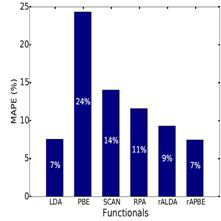
Figure 3 (left): Surface energies averaged over the three lowest energy crystal faces are compared against the experimental energies.
Figure 4 (right): Overall mean absolute percentage error (MAPE) in the surface energies of functionals. Relative error for the surface energies is calculated from the average value of the
surface energies of (111), (110) and (100) surfaces of the metals and compared to measured liquid-metal surface tensions extrapolated to 0 °K.
We always need a beyond-RPA treatment for bulks, but a properly constructed kernel does not strongly affect the surface energy. The kernel contribution tends to reduce the overestimation of the surface energies of RPA, yielding quite good results on average. Kernel corrections systematically improve the short-range correlation of RPA and boost its accuracy for surface energies.
The work on Renormalized RPA resulted in one publication (and an erratum with the proper form of acknowledgment has been submitted). A second publication regarding the results on metallic and semiconducting surface energies is expected in the close future.
2. Adsorption of organic molecules on metallic and semiconducting surfaces: The importance of the vdW contribution to surface properties has been emphasized recently in the work of Patra et al. (PNAS, to appear). By naturally accounting for both intermediate and long-range interactions, the recently developed SCAN+rVV10 approximation represents a major improvement over previous functionals for many properties of diversely-bonded systems. Even with current computational power, the RPA-based methods are expensive. In practice, especially when we want to evaluate adsorption energies, we need an approximation which is comparable in its accuracy but comes at a more reasonable cost. This is exactly what SCAN+rVV10 can provide us. The justification of using the SCAN+rVV10 method then relies on both the works of Patra et al., and our results on RPA.
Currently we are testing the SCAN and SCAN+rVV10 approximations for the adsorption of organic compound to semiconducting surfaces. The adsorption of cyclic organic molecules is crucial in the hydrodesulfurization (HDS) catalysis. In our assessment we include thiophene and benzothiophene. Additional compounds may include butadiene, pyridine, quinoline and naphthalene.
Since our work focuses on vdW bonding between the organic compound and the substrate, we can follow either the DDS or HYD pathways on the so-called S and Mo sites respectively, as described in Moses at al., Journal of Catalysis 248 (2007) 188. At first we choose the S edge site. We investigate various adsorption configurations (in the case of thiophene and its pre-hydrogenated form as examples). We are applying the SCAN meta-GGA and the SCAN+rVV10 vdW corrected meta-GGA for the exploration of vdW interactions in the adsorption process.
We are extending our understanding of the adsorption process toward metallic surfaces. The adsorption of organic compounds on metallic surfaces is particularly challenging for vdW approximations. References claim that in this case the non-additive many-body effects can be significant. The adsorption of thiophene is currently being assessed on a Cu surface for all the lowest energy crystal faces.










