Reports: DNI352452-DNI3: A Nonmetal Platform for Inner Sphere Two-Electron Redox Catalysis
 printer friendly
printer friendly
Figure 1. Catalytic hydrogen transfer reactivity of 1/[H]2.
In this funding year, we have demonstrated that [H]2 is capable of transferring hydrogen-equivalents to substrates other than azobenzenes, including aldimines, ketimines, and certain highly electron deficient alkenes. We have not observed on reaction (hydrogen transfer or otherwise) between [H]2 and less activated alkenes or alkynes.
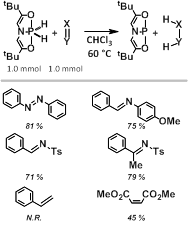
Figure 2. Hydrogen transfer of [H]2 to unsaturated substrates.
We have further investigated the formation of mixed isotopologues of [H]2. In analogy to the preparation of [H]2 by dehydrogenation of ammonia-borane, treatment of 1 with D3NBD3 results in conversion to the dideuteriophosphorane [D]2. Attempts to prepared the mixed isotopologue [H][D] by an analogous procedure with D3NBH3 have failed. Instead, [D]2 is the sole phosphorus containing product obtained from this reaction, isolated in good chemical yield. Complementarily, when H3NBD3 is employed, only [H]2 is obtained. In short, it is the nitrogen substituent that governs the outcome of the mixed isotopic experiments. We believe this to be function of facile P–H exchange between the pentacoordinate phosphoranes and the labile N–H(D) of the ammonia-boranes. Specifically, [H]2 can be converted to [D]2 by treatment with excess D3NBH3 but not H3NBD3. Indeed, a similar isotopic exchange can be observed upon treatment of [H]2 with excess BnND2.
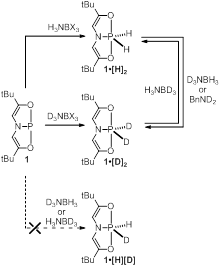
Figure 3. Synthetic routes to [H]2 and [D]2 by dehydrogenation of ammonia-borane and isotope exchange.
The 1H NMR spectra of [D]2 is identical to the spectra for [H]2 except for the absence of signals for the hydrogen isotopes bonded to phosphorus. The 31P NMR signal for [D]2 is shifted to -46.3 ppm, as compared to -45.3 ppm for [H]2. The 31P{1H} NMR provides a quintet formed from the coupling of phosphorus with two equivalent 1 2H nuclei (S=1), such that 1JP-D= 103 Hz. In the 1H-coupled 31P NMR the signal forms a quintet of triplets, with a 3JP-H= 34 Hz; the magnitude of coupling with the vinylic protons of the ligands is unaffected by the isotopic substitution. In the 2H NMR the 2H nuclei are observed as a doublet at 8.03 ppm with a 1JP-D= 103 Hz. It is notable that no other 2H signals were observed, indicating that no deuterium incorporation occurs at the vinylic position. While [H][D] cannot be obtained cleanly, it is observed as an intermediate in the formation of [D]2 from [H]2. [H][D] is also obtained upon mixing [D]2 from [H]2 in benzene-d6. The isotope exchange takes place after 12 h at room temperature, or within an hour at 60 °C to provide a statistical mixture of the three isotopologues. [H][D] is characterized by a doublet of triplets of triplets at 31P NMR d=-45.8 ppm, a 1JP-D= 103 Hz, a 1JP-H= 316 Hz and 3JP-H= 34 Hz.
Support from the ACS-PRF over the course of the two-year funding period has had significant impact on the success of this research, and has had significant positive influence on the training of graduate students. A talented Ph.D.-candidate (Nicole Dunn) was supported by this award; she is now nearing completion of her thesis that will be based in large part on the results obtained through ACS-PRF support. In addition to those already published, several additional manuscripts acknowledging ACS-PRF support are anticipated in the near term. The support provided by the ACS-PRF DNI award has been instrumental in obtaining preliminary data sufficient to compose successful grant applications for renewable federal funding, enabling continued investigation of this and related main group systems. Additionally, due in part to the research supported by the ACS-PRF, the PI has been generously recognized with several young faculty awards.










