Reports: DNI1052404-DNI10: Validating Computational Design Principles for Crystalline Enzyme Assemblies
 printer friendly
printer friendlyMonomer Redesign for Solubility
Optimizing Adventitious Crosslinking
Efficient Crystal Search Algorithm in Hexagonal Space
Groups
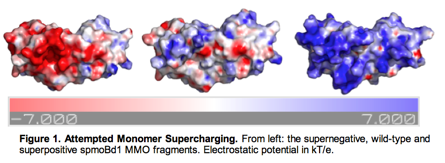
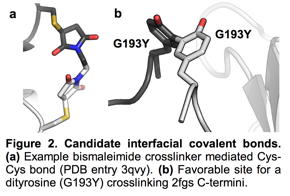
Monomer Redesign for Solubility
Optimizing Adventitious Crosslinking
Efficient Crystal Search Algorithm in Hexagonal Space
Groups










