Reports: ND652100-ND6: Multiscale Investigation of Asphaltene Self-Assembly
 printer friendly
printer friendlyAsphaltenes form the heaviest and most aromatic fraction of
crude oil. There exist two dominant paradigms for developing CG models
of liquids and other soft materials. In the second funding period, we extended and applied this
methodology to parameterize transferable bottom-up CG models that not only
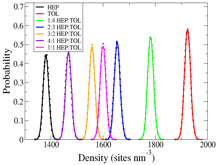
accurately model the equilibrium structure, but also the density and
compressibility of hep-tol mixtures.
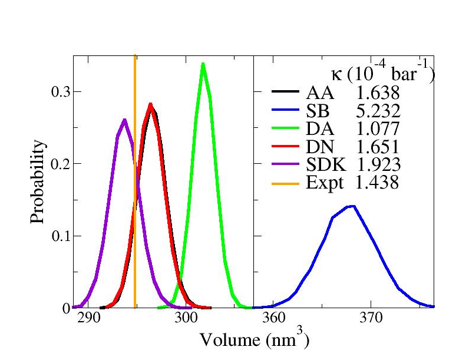
In particular, the above figure compares the experimental (orange)
density and compressibility with the volume distributions and compressibilities
generated by the atomically detailed OPLS model (black) and by several 3-site CG
models for heptane. In addition, we have applied this method to parameterize
volume-dependent corrections to the virial for application with transferable
potentials for hep-tol mixtures.
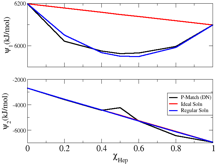
Furthermore, we have also worked on developing a top-down (thermodynamics-based)
CG model for hep-tol mixtures.
Finally, we have also begun work on parameterizing CG models for model
asphaltene molecules.