Reports: ND351715-ND3: A New Method to Link Metal Ions via Cycloaddition of Metal-Azides to Metal-Acetylides
Adam Veige, PhD, University of Florida
The
goal of this grant was to develop a deeper understanding of the 1,3-dipolar cycloaddition reaction between metal-azides and
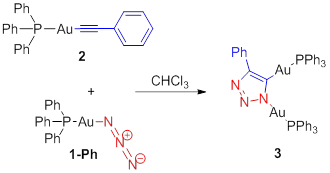
metal-acetylides, a transformation we have labeled as iClick.Our preliminary results (Scheme 1) have shown
that gold(I) is amenable to this type of reaction, as demonstrated by the
reaction of triphenylphosphinegold(I) azide (1-Ph) and triphenylphosphinegold(I)
phenylacetylide (2),
which undergo a 1,3-dipolar cycloaddition reaction to yield the triazole bridged product,
4-phenyl-1,5-triphenylphosphinegold(I) 1,2,3-triazolate (3). As outlined in the
proposal, we planned to probe the scope of the iClick reaction, in terms of
metal compatibility, ancillary ligand effects, and acetylene substituents.
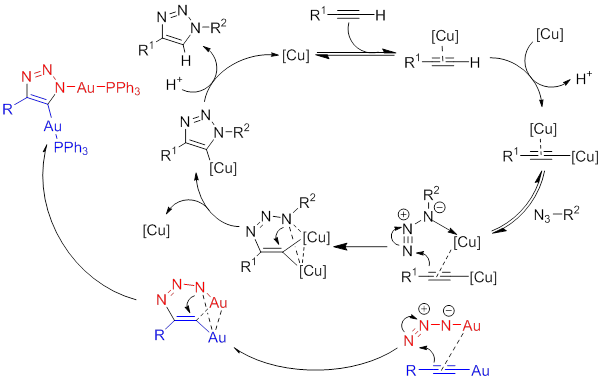
A series
of kinetic experiments were designed to probe the mechanism of the iClick
reaction between a variety of gold(I) azides and
gold(I) acetylides.The reaction proved
to be first order in both reactants, or second order overall, with respect to gold(I) concentration.
This was not surprising, as the generally accepted mechanism of Copper(I)-catalyzed Azide Alkyne Cycloaddition reactions (CuAAC) is also second order with respect to the copper
concentration, a first row congener of gold. The proposed mechanism is depicted
in Scheme 2, alongside the proposed CuAAC mechanism. The necessity for ligand dissociation was confirmed
by a significant rate inhibition when free triphenylphosphine was
systematically added to the reaction.
This finding is further corroborated by results indicating
N-heterocyclic (NHC) supported gold(I) azides and acetylides
to not proceed under ambient conditions; as NHC ligands are stronger
σ–donors than phosphine ligands, it is harder for this crucial ligand
dissociation to occur. Lastly, when the
para-proton of the phenyl acetylene was systematically substituted with both
electron withdrawing (NO2 and F) and electron donating (OMe) groups, the gold(I)/gold(I)
iClick reaction rates followed a linear Hammet plot,
with a slope of 0.976. This slope
indicated there is a significant buildup of negative charge in one of the
intermediates, which is stabilized by the more electron withdrawing
substituents, leading to faster reaction rates.
A nucleophilic attack of the terminal nitrogen of the azide by the b-carbon of the acetylide, could
indicate a reasonable location of the partial negative charge in the
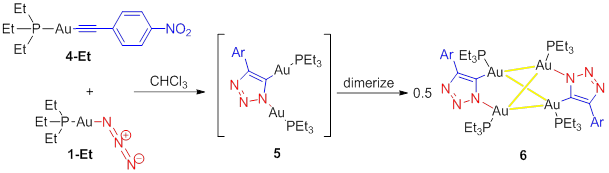
intermediate. When
the triphenylphosphine ligands are substituted with the more basic, yet
conically smaller triethylphosphine ligands (1-Et and 4-Et), the reaction is effected notably.While the reaction proceeds
to a similar triazolate-bridged, dinuclear intermediate (5), strong intermolecular aurophilic interactions cause a
dimerization (6) to occur (Scheme 3). This dimerization is permitted by the less
sterically demanding triethylphosphine ancillary
ligands. The crystallographically
characterized product consists of a pseudo C2-symmertic
distorted tetrahedral geometry of gold(I) ions, and
can be seen in Figure 1.
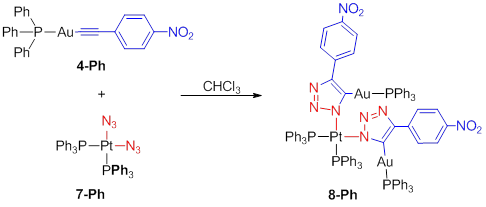
To
demonstrate the compatibility of other metals in an iClick type reaction, a
series of phosphine supported platinum(II) di-azide
complexes were synthesized.Triphenylphosphine supported platinum(II) diazide7-Ph
readily undergoes two subsequent iClick reactions with two equivalents of gold(I)
acetylide 4-Ph. When this reaction is carried out in CDCl3,
the stepwise iClick reactions can be monitored over a 12 hour period, but when
run in C6D6, at slightly elevated temperatures, the
desired, trinuclear product (8-Ph) crystalizes
out in 92% isolated yield.
The
effect of the ancillary ligand was monitored, and once again,
triphenylphosphine was substituted with triethylphosphine.While the desired trinuclear product 8-Et was isolated, the yield of the
reaction suffered from deleterious side-reactions, which were the result of
anionic ligand exchange of the reactants.
While anionic ligand exchange was not observed when triphenylphosphine
was utilized, the more strongly σ–donating triethylphosphine
located trans
to the azido functionalities leads to increased labilization. The other two isolated products, were the
gold(I)/gold(I) iClick product, 6,
and 9, the result of a single iClick
reaction between one of the azides on the platinum(II), and the acetylide of
the gold(I), followed by anionic ligand exchange of the other azide with an
acetylide from another gold(I) (Scheme 5).
Multinuclear and 2-D NMR spectroscopy data provides reasonable evidence
for the identity of 9, though its
isolation from the product mixture was not possible.
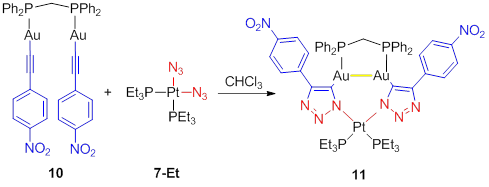
To
create a complex in which an gold(I)-gold(I)
interaction could be manipulated, we sought to forcibly constrain the gold(I)
ions in close proximity. Tethering the gold(I) acetylide units across a bis(diphenylphosphino)methane (dppm)
bridge (10) provides the necessary
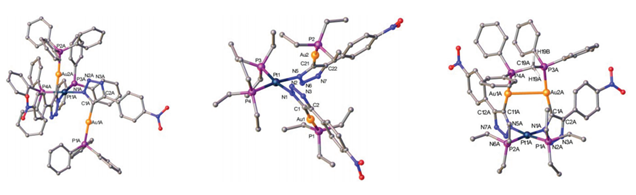
structural constraint, and allows the synthesis of the trimetallic PtII/AuI2 complex 11 in 36% yield (Scheme 6). Figure 2
shows the solid state structure of 8-Ph,
8-Et, and 11, with hydrogen atoms and disordered atoms removed for
clarity.Both pseudo-C2-symmetric, square planar
complexes, 8-Ph and 8-Et, the gold(I) ions orient anti, relative to each other, but the C1-symmetric 11, features gold ions oriented in a syn fashion, with an aurophilic
interaction between them (Au-Au bond distance of 3.1878(10) Å).
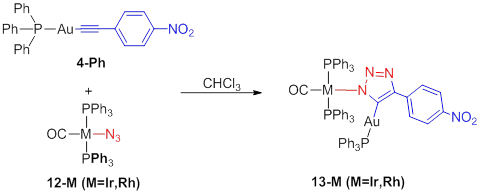
We also
attempted to employ the second row palladium congener of 7, however the reaction results
in an intractable mixture of products.Both third and second row iridium(I) and rhodium(I) azides do
participate in iClick with gold(I) acetylides, as depicted in Scheme 7. These products have been characterized by NMR
spectroscopic techniques.
In
conclusion, this grant has supported the research to better understand the
scope of the iClick reaction. We now have a firm understanding of the reaction
mechanism, but more importantly, we now understand the limitations of the
iClick reaction. This project, as is its intention, is to nurture new research
directions. The results from this project were used as preliminary result to
obtain additional support from the DOE-BES materials division. The project was
initiated by one undergraduate student (Trevor Del Castillo) who went on to win
the prestigious NSF graduate fellowship and is now pursuing his PhD at Cal Tech
under the guidance of Prof. Jonas Peters. Currently three graduate students are
working on this project within the Veige laboratories.Andrew Powers is
scheduled to graduate with a Ph.D. in the spring of 2015. Xi Yang (Ph.D., 2016)
and Chris Beto (Ph.D., 2018) will continue the project
Scheme
1
Scheme
2
Scheme
3
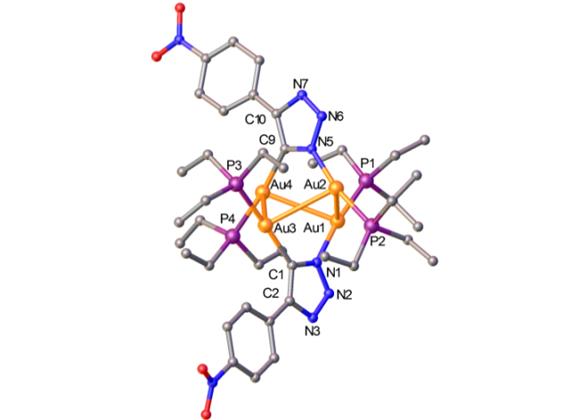
Fig.
1.Solid state
molecular structure of 6.Scheme
4
Scheme
5 Scheme
6Fig. 2Solid state structures of 8-Ph (left), 8-Et (center) and 11
(right).
 printer friendly
printer friendly