Reports: DNI353536-DNI3: Frustrated Metal-Ligand Interactions: C-H Activation and Functionalization
 printer friendly
printer friendly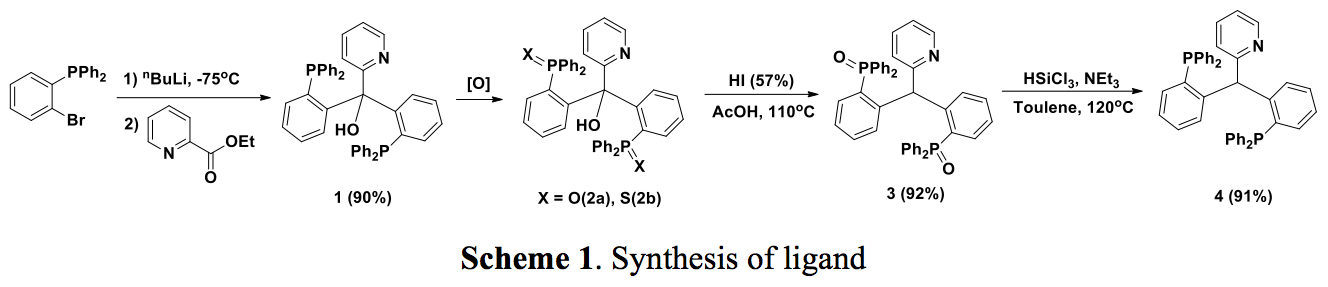
The synthesis of pyridine-based PCpyP-pincer ligand was slightly different, key reactions including a protection of phosphine 1 to the corresponding phosphine oxide (2a) or sulfide (2b) prior to the reduction of the alcohol group. The phosphine sulfide 2b was directly hydrolyzed to the phosphine oxide 3 under this reaction condition. Reduction of phosphine oxide 3 with HSiCl3 in the presence of base afforded the desired compound 4 (Scheme 1).
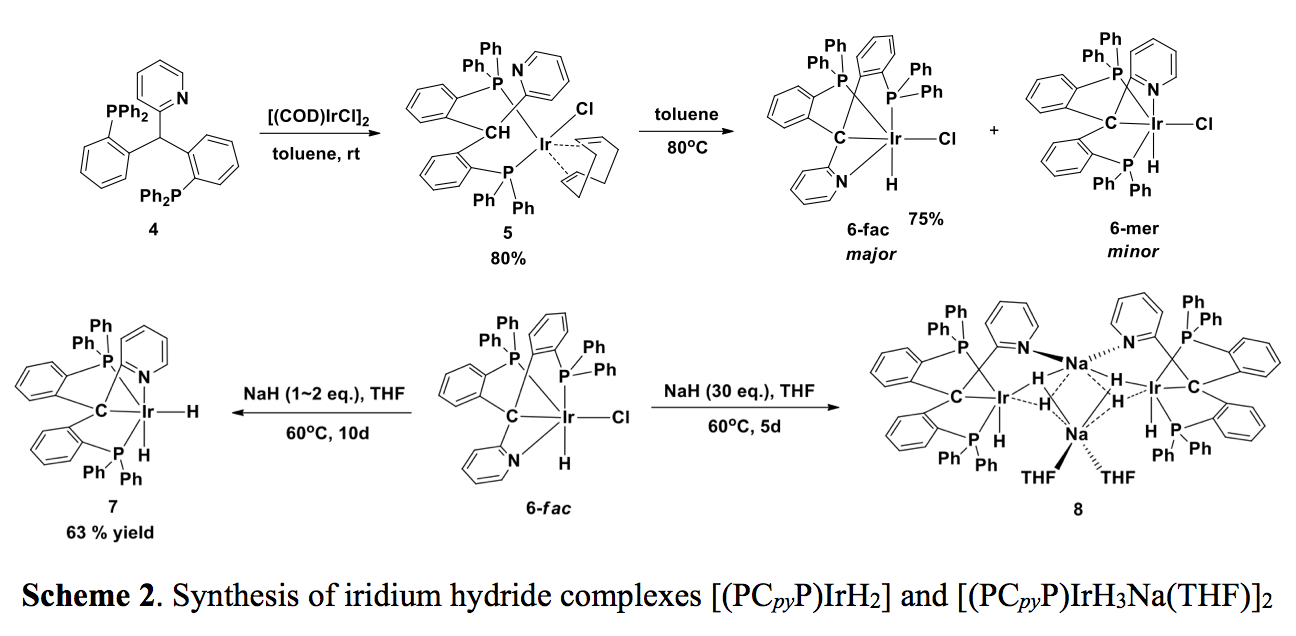
Reaction of 4 with [(COD)IrCl]2 at room temperature initially affords a ?-2 adduct, 5, which was isolated (Scheme 2). 5 is stable as a solid, but when heated in toluene two iridium chloride complexes, 6-fac and 6-mer, were formed, with the facial isomer as the major product. 6-fac can be isolated as a pale-yellow solid in 75% yield by recrystallization from CH2Cl2/ether. Different reactivity was observed when the more labile [(COE)2IrCl]2 reacted with 4: only 6-fac was formed and no other intermediates were observed by NMR spectroscopy.
The iridium chloride complex 6-fac reacted with NaH in THF to give the iridium dihydride complex 7 (Scheme 2). Interestingly, when a large excess of NaH reacted with 6-fac, a dimeric iridium trihydride complex 8 was formed that contains two Na ions bridged by hydrides. The crystal structure of 8 showed that one Na ion was coordinated by two pyridine groups of the ligands, while the other Na ion coordinated two THF molecules.
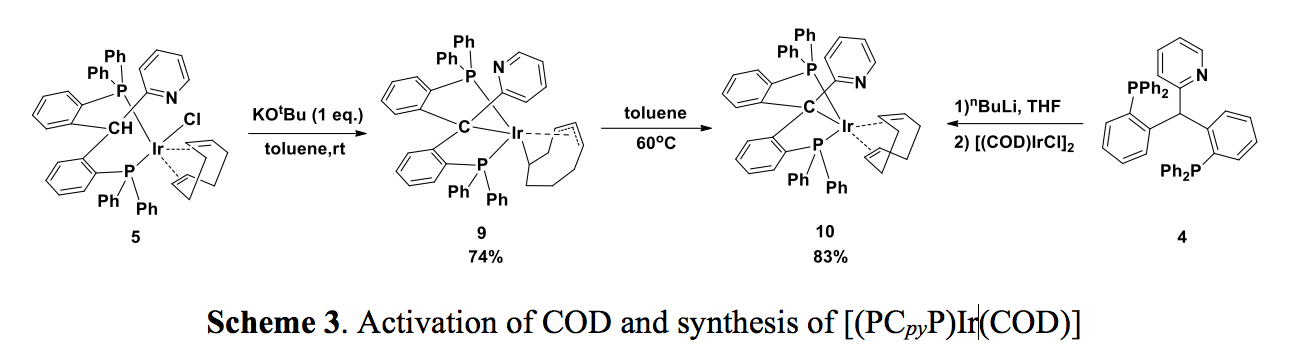
Iridium(I) complexes can serve as reactive catalysts, therefore, we also tried to synthesize the corresponding iridium(I) complexes supported by 4 (Scheme 3). Treatment of the ?-2 adduct 5 with KOt-Bu at room temperature afforded 9. Though an Compound 9 features a dianionic COD ligand, formed by the activation of the neutral COD ligand at the allylic position, which generated in-situ an iridium(I) species, followed by insertion of the other C=C bond of the COD ligand into the Ir-H bond. Upon heating of 9 in toluene, the expected iridium(I) complex 10 was formed as the major product. Complex 10 can also be synthesized by the deprotonation of 4, followed by a salt metathesis reaction with [(COD)IrCl]2. A variable temperature NMR study (25 ~ 80 oC) of 10 indicated a hindered rotation of the pyridine group; coalescence was observed in the 31P NMR spectrum at 65 oC. The activation barrier was also estimated (ΔG0 = 66.5 kJ/mol, ΔH0 = 76.04 kJ/mol and ΔS0 = 35.75 J/moláK) using an Arrhenius plot.
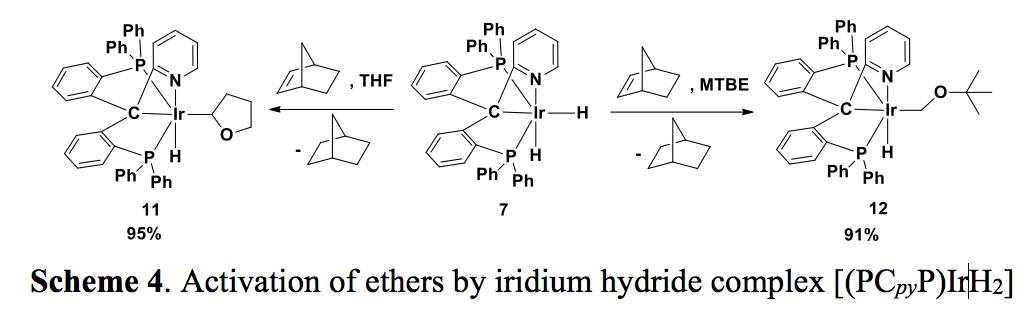
Initial reactivity studies showed that neither the iridium chloride 6-fac nor the dihydride 7 are good catalysts for alkane dehydrogenation under various conditions, probably due to the relatively strong coordination of the pyridine group in nonpolar reaction media such as cyclooctane and tert-butylethylene. However, we found that the dissociation of pyridine is more favorable in a more polar solvent, and C-H bond activation reactions are facilitated. Therefore, upon treatment of 7 with norbornene (NBE), both THF and MTBE can be selectively activated at their a-positions to give the corresponding hydride complexes 11 and 12 in high yields (Scheme 4).
Interestingly, heating of 11 or 12 did not afford a Fischer-carbene complex through a second C-H bond activation. Instead, the dihydride complex 7 was quantitatively formed when 11 was heated in THF, with 2,3-dihydrofuran likely being the byproduct from the ß-H elimination. On the other hand, heating 7 in benzene generated [(PCpyP)Ir(H)Ph] as the only iridium species, is a consequence of the C-H bond activation of the solvent; free THF was also observed. These observations indicated that in a more polar solvent or in the presence of polar reagents, the dissociation of the pyridine group is facile, explaining the ß-H elimination process in the case of THF and the sequential reductive elimination / oxidation addition process in the case of benzene. Since 12 has no ß-H available, the reductive elimination of free MTBE was observed, and [(PCpyP)Ir(H)Ph] was formed from benzene.
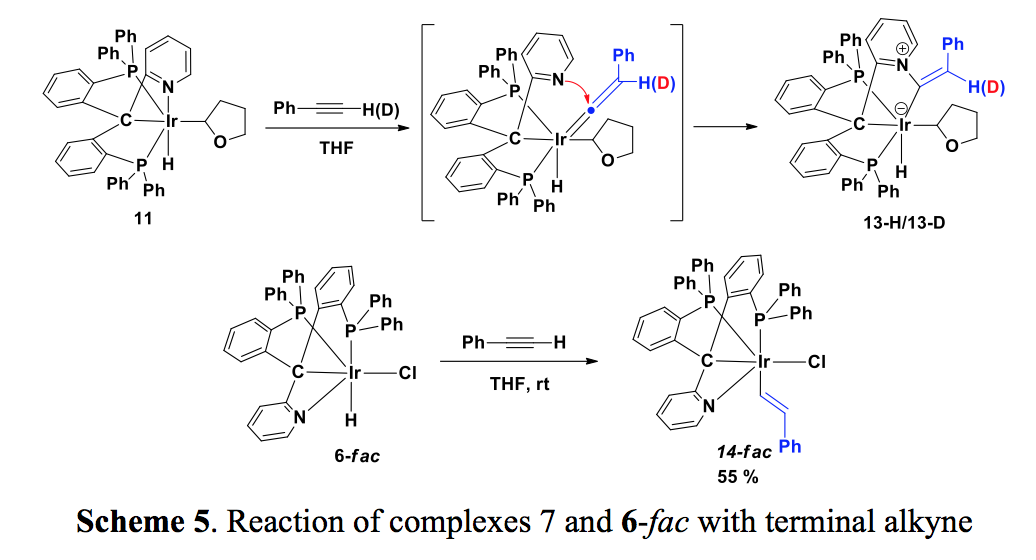
Although the coordination of the pyridine group to iridium blocked the second C-H bond activation, it helped to stabilize 11 and 12. Considering the dissociation of pyridine group in the presence of polar reagents and its basic nature, we tried to functionalize the THF group in 11. Treatment of 11 with phenylacetylene afforded a zwitterionic compound, 13, which was isolated and characterized by X-ray diffraction studies. In 13, the original THF group and the hydride are still intact; attack of the pyridine group to a vinylidene ligand resulted in the formation of an olefinic substituted pyridinium ligand (Scheme 5). The formation of the vinylidene intermediate is supported by using phenylacetylene-d1 (Scheme 5, 13-D). The isolation of 13 indicates that the pyridine group may be involved in certain reactions due to its basic and nucleophilic nature. The reaction of 6-fac with phenylacetylene afforded the expected Ir-H bond insertion product 14, in which the fac configuration was retained (Scheme 5).
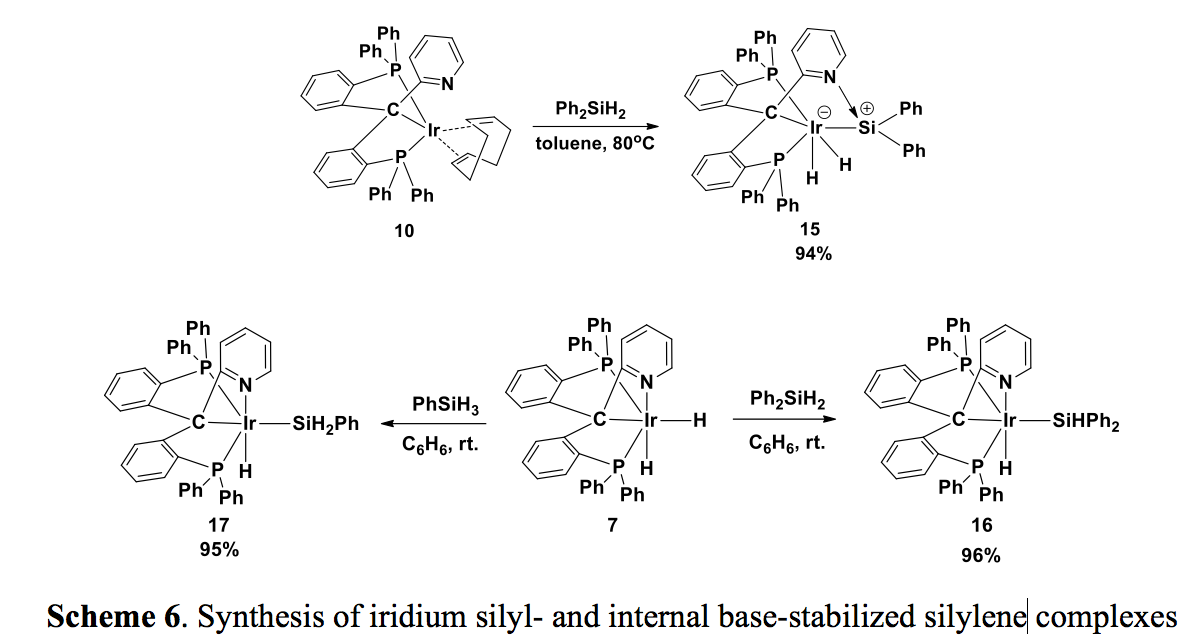
The iridium(I) complex 10 is potentially a good starting material for a range of oxidation addition reactions. However, 10 does not react with alcohols at elevated temperature. No reaction was observed with phenylacetylene. Instead, 10 slowly reacted with Ph2SiH2 to give an interesting iridium silyl dihydride compelx 15 in high yield (Scheme 6). The 29Si NMR spectrum showed a resonance at 47.6 ppm, which results from coupling of the 29Si nucleus with both cis (9.8 Hz) and trans (172.1 Hz) phosphines, significantly upfield shifted compared to corresponding signals in base free iridium silylene complexes (~250 ppm). Based on this spectroscopic data, we propose that complex 15 is an internal base stabilized iridium silylene complex. Recently, we found that the dihydride complex 7 reacts with both PhSiH3 and Ph2SiH2 under mild conditions to give the corresponding iridium silyl hydride complexes 16 and 17 in high yield. We are currently studying the conversion between these iridium silyl and silylene species.










