Reports: UR353427-UR3: Homogeneous Fischer-Tropsch Catalysts for the Conversion of Syngas into Higher Order Hydrocarbons
John D. Gilbertson, PhD, Western Washington University
The predicted decrease in the
availability of petroleum and petroleum-based products has sparked renewed
interest into the conversion of synthesis gas (syngas) into chemical feedstocks and fuels. Conversion of syngas (CO + H2)
to liquid hydrocarbons and a-olefins by the Fischer-Tropsch (F-T) reaction offers promise, but the low
selectivity of these reactions by the heterogeneous route is a major
drawback. The development of homogeneous systems for the F-T reaction has
the potential to increase both selectivity and understanding of the F-T
mechanism. Funded by an Undergraduate Research grant from the American
Chemical Society Petroleum Research Fund (UR, June 2013 – August 20116),
metal-ligand complexes composed of pendant Lewis/Bronsted
acids/bases and redox-active sites within the ligand scaffold are being
investigated to study the C-H and C-C bond forming steps in the F-T reactions
of CO. To date, we have successfully
synthesized a series of novel metal-ligand complexes based on the pyridinediimine (PDI) scaffold that contain pendant amine
bases (ABPDI), pendant Lewis acids (LAPDI), or frustrated
Lewis pairs (FLPPDI) located in the secondary coordination
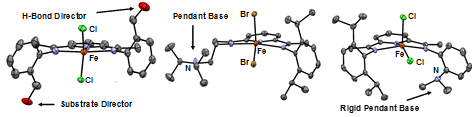
sphere. A handful of these new complexes are shown in Figure 1.
Figure 1. ORTEPs
of representative, unique PDI complexes with reaction directors located in the
secondary coordination sphere synthesized and outlined in this progress report.
One
of the major goals outlined in the original proposal was to utilize the
secondary coordination sphere to stabilize the formation of CO-derived iron formyls (formed from the addition of hydride to the reduced
dicarbonyl species). However, reduced dicarbonyl species formed from the complexes in Figure 1 do
not display any formyl formation. Instead, deprotonation of the –CH3 groups on the 2,6-diacetylpyrindine core occurs upon the addition of
hydride due to the acidity of the acetyl protons in these complexes. In
order to circumvent these unwanted side reactions, we have begun to synthesize
complexes based on the 2,6-dibenzoylpyridine core
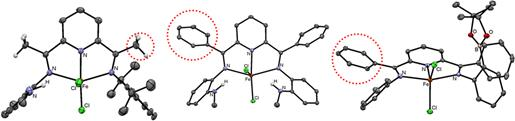
instead. Examples of these complexes are shown in Figure 2.
Figure 1. ORTEPs
of representative FePDI complexes highlighting the
acidic –CH3 (left) and the nonacidic –C6H5
groups (middle and right) in the PDI backbone.
We
are currently exploring the reduction chemistry of these compounds by producing
reduced iron dicarbonyls.We
are also in the process of synthesizing tripodaltetraamine-based ligands with pendant Lewis/Bronsted acids/bases and redox-active sites within the
ligand scaffold. The tripodaltetraamine fragment is a common building block in inorganic
chemistrydue to its ability to stabilize metal complexes with uncommon
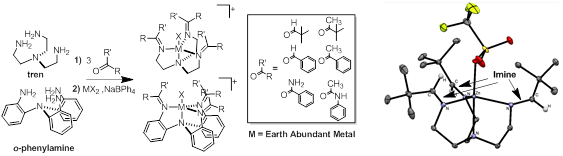
geometries and hence diverse catalytic capabilities. As shown in Figure
3, we are investigating the tripodal systems based on
the tren backbone and also the rigid o-phenylamine backbone. We are integrating redox active
sites into these systems by synthesizing the Schiff base analogs of the tren and o-phenylamine
backbones. These systems have potential advantages over the PDI systems
described above. It is likely that in the COreduction
reaction that multiple electron equivalents may be stored within these
scaffolds while still only chelating one CO molecule (instead of two in the PDI
system). The o-phenylamine backbone has
been shown to form ligand-based radicals upon oxidation but the reduction
chemistry is largely unexplored. We have successfully synthesized the
zinc triflate analog of the tris-tert-butylimine
complex based on the tren core, as shown in Figure 3
(right).
Figure 3. Synthesis of tripodal systems with reaction directors and redox active
sites incorporated into the ligand scaffold.
In the next year we
are planning to publish our new complexes, as well as continue to investigate
the reactivity of these complexes in the C-H and C-C bond forming steps in the
F-T reactions of CO.
 printer friendly
printer friendly