Reports: DNI652494-DNI6: Dynamics and Kinetics of Carbon Dioxide in Nano-Porous Materials Environment from Quantum Molecular Dynamics Simulations
Yosuke Kanai, PhD, University of North Carolina (Chapel Hill)
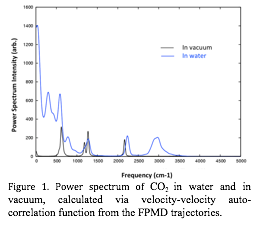
Figure 1. Power spectrum of CO2 in water and in vacuum, calculated via velocity-velocity auto-correlation function from the FPMD trajectories.
Several key progresses were made in
few different areas during the second
year of the grant period (9/1/2013-8/31/2014).
CO2 in H2O
We have
been investigating how water solvation influences the CO2 dynamics. As
the first step, we have calculated the vibrational spectrum of CO2 in
vacuum and in water. From the trajectories of first principles molecular
dynamics (FPMD), the power spectrum can be obtained by taking the Fourier
transform of the velocity-velocity auto-correlation function between atoms. In
vacuum, the well-known splitting at around ~1300 cm-1 due to Fermi resonance is
apparent (Figure 1). However, with water solvation, much of the detailed
features are washed out, and there is a prominent broad peak centered around 3000 cm-1. This broad peak was identified to derive
from the hydrogen bonding with surrounding water molecules.
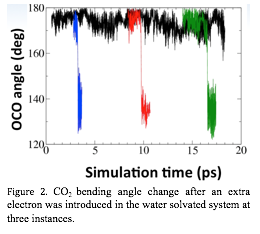
We also
started to investigate how the presence of a solvated electron influences the
dynamics of CO2 in
water. From the trajectory of the neutral CO2 in water, we arbitrarily
selected three configurations (separated by the minimal of 2 picosecond) and
re-optimized the electronic structure of the system with an extra electron
before proceeding with the molecular dynamics. The CO2 bending
angle was monitored during the simulation, and the initially fluctuation of CO2 angle
around 170¡ was
quickly and rather abruptly reduced and began fluctuating around 135¡ in all three cases (Figure
2).
Figure 2. CO2 bending angle change after an extra electron was introduced in the water solvated system at three instances.
H2O in confinement
FPMD
simulations were used to gain an atomistic-level insight into how the molecular
behavior of interfacial water is influenced by specific surface adsorbates. Although the overall hydrophobic versus
hydrophilic character of a given surface is widely recognized to be important
in determining the behavior of interfacial water molecules, we have shown that
subtle molecular details may also play a role in determining the dynamical
behavior of water. By comparing water diffusivity at three different nonpolar
surfaces, we find that specific surface features can lead to a suppression of
hydrogen bond network ring structures by enhancing hexagonal spatial
distributions of water molecules near the surface. Such a distinct
molecule-dependent behavior of the interfacial water was found to persist well
into the liquid, while most structural properties are noticeably influenced in
only the first water layer.
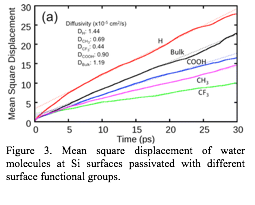
In
particular, water molecules diffuse significantly faster at the H−Si
surface than at CH3−
or CF3−Si
surfaces because the hydrogen bond ring network formation is suppressed at
those surfaces (Figure 3). The electrostatic potential generated by the adsorbate species appears to dictate the spatial water
distribution in the first water layer. Such behavior is clearly noted for both
metal- and carbon-based surfaces in the literature. Interestingly, such a distinct
long-range molecular behavior in the first water layer extends farther away (∼1 nm) because HB ring
sizes
Figure 3. Mean square displacement of water molecules at Si surfaces passivated with different surface functional groups.
are appreciable (4−5) in water. Our
present study shows that long- range structural arrangements of water molecules
are also important to dictate the dynamical behavior of water molecules at
surfaces passivated by different hydrophobic
molecules. The observed structural dependence on the dynamics of interfacial
water is likely to be a universal character for a wide range of surfaces.
Role of Charge
Transfer in describing Water Diffusivity
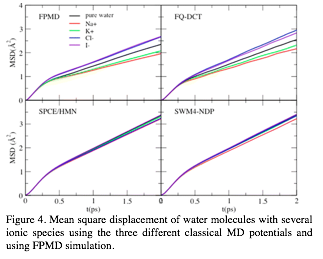
Figure 4. Mean square displacement of water molecules with several ionic species using the three different classical MD potentials and using FPMD simulation.
During the investigation of the CO2
hydrolysis to bicarbonate ion in bulk water, it has become clear that classical
molecular dynamics simulations are necessary to address some important
dynamical properties such as diffusion constant of the water molecule when CO2
molecule is solvated. First-principles molecular dynamics (FPMD) turns
out to be computationally too expensive for computing the diffusion constant
accurately in terms of statistical sampling. At the same time, most classical
molecular dynamics is known to suffer from the inadequate treatment of charge
transfer. Building on the recent work by Rick and co-workers (1), we
investigated the performance of the new potential that can take into account
both electronic polarization and charge transfer. Classical molecular
dynamics simulations were performed using three different force fields: a fixed
charge force field (SPCE/HMN), a polarizable force field that includes explicit
polarization (SWM4-NDP), and also a recently developed force field that
includes polarization and charge transfer (QF-DCT). These simulations were then
compared to FPMD simulations. We performed molecular dynamics simulations on
four types of systems containing ion and solvating water. Two systems contained
a cation (Na+ or K+), and two other systems an
anion (Cl– or I–) as representative cases.
While the first-principles simulations showed that the anions accelerated water
translational diffusion, the cations slowed it down.
In simulations with the classical force fields, only the force field that
incorporates explicit charge transfer reproduced this ion-specific behavior
(Figure 4). Additional simulations performed to understand the effect of charge
transfer demonstrated that two competitive factors determine the behavior of
water translational diffusion: the ions diminished charge accelerates water,
while the net charge acquired by water either accelerates or slows down its
dynamics. Our results showed that charge transfer plays a crucial role in
governing the water dynamics in aqueous ionic solutions.
References
1. Soniat, M.; Rick, S. W. The
Effects of Charge Transfer on the Aqueous Solvation of Ions.J. Chem. Phys. 2012, 137 (4), 044511.
 printer friendly
printer friendly