Reports: UR151961-UR1: Development of a Catalytic, Asymmetric Aza-Cope Rearrangement and Mannich Cyclization
 printer friendly
printer friendly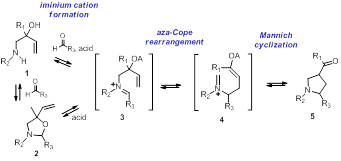
[1] Overman, L. E.; Kakimoto, M.-A. J. Am. Chem.
Soc. 1979, 101, 1310-1312.
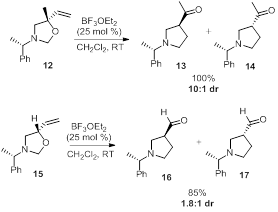
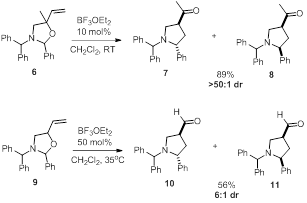
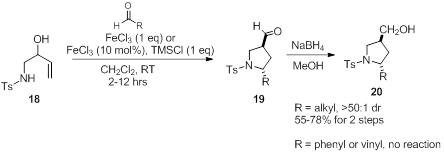
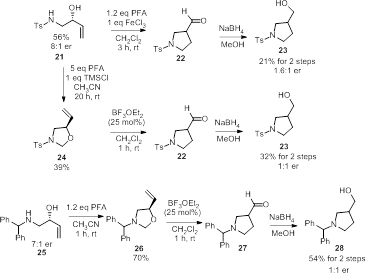
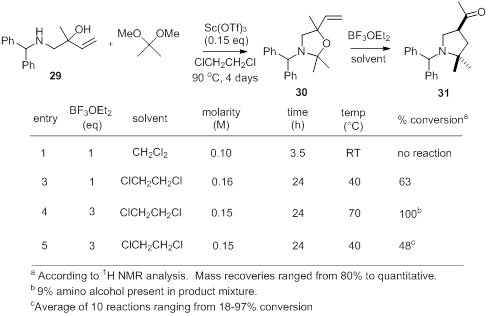
[1] Overman, L. E.; Kakimoto, M.-A. J. Am. Chem.
Soc. 1979, 101, 1310-1312.










