Reports: DNI151975-DNI1: Stereoselective Homoallylation of Aldehydes and Related Compounds
 printer friendly
printer friendlyThe goal of the proposed research is to develop reagents and
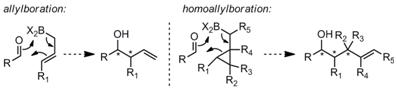
catalysts for stereoselective homoallylation and homocrotylation (Scheme 1). In
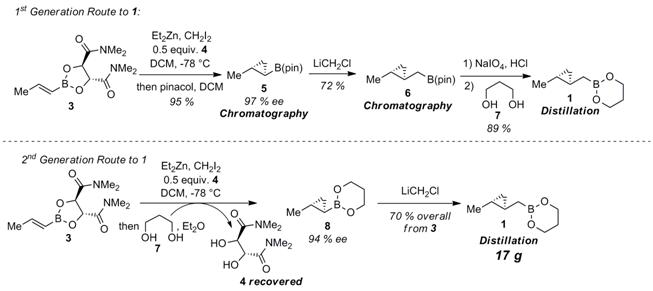
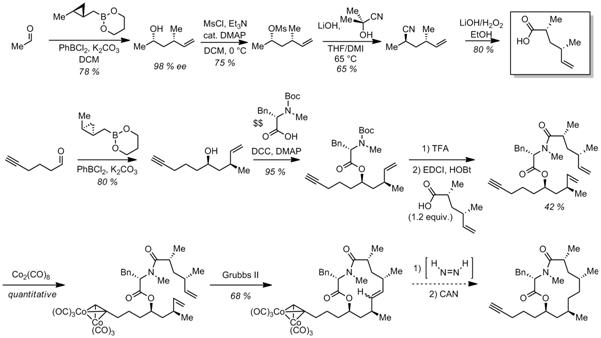
the second year of this grant, we have made progress in 1) asymmetric Scheme 1. First and Second-Generation Routes to
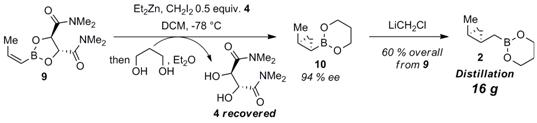
Scheme 2. Second-Generation Route to
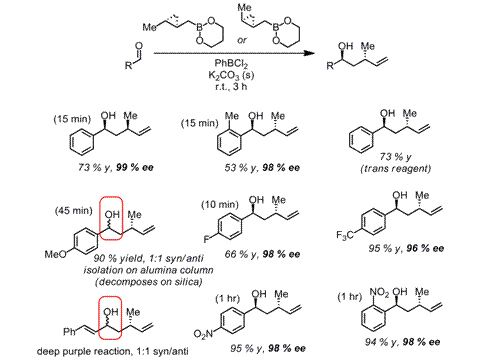
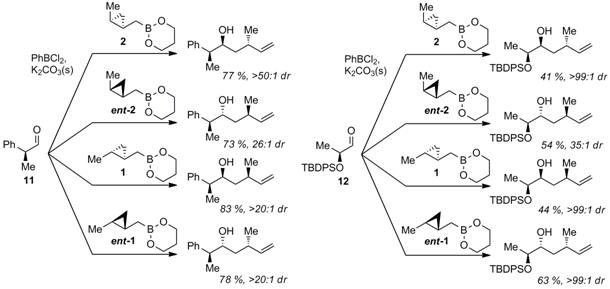
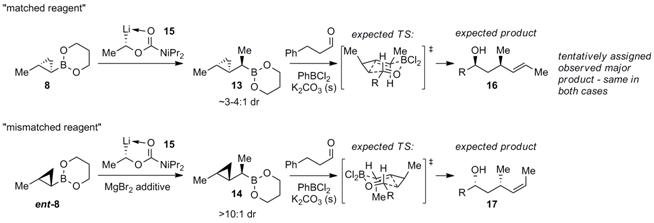
We have also extensively explored double diastereoselection
with these reagents. Selected examples are shown in Scheme 4, in which we
manage to obtain all possible stereotriads in adducts
to chiral aldehydes, containing either all-carbon stereocenters (