Reports: DNI353621-DNI3: Redox Robust Non-Noble Metal Catalysis
Jeffery Allen Byers, PhD, Boston College
During this funding period, we have extensively studied the
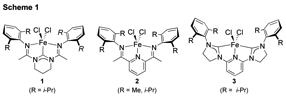
coordination chemistry, bonding, and catalytic properties of bis(amidinato)-N-heterocyclic carbene iron complexes (e.g. 1, Scheme 1), which are structural
analogs to the catalytically prolific bis(imino)pyridine
iron complexes (e.g. 2, Scheme 1).{JACS1999, 121, 8728; JACS 1998, 120, 4049; JACS 2006, 128, 13340; Science 2012, 335, 567} In addition to these
studies, we have initiated a program dedicated to developing iron-catalyzed
cross coupling reactions using iron complexes (e.g. 1-3, Scheme 1) containing redox active ligands.
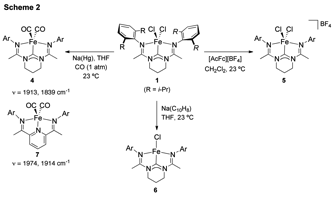
By carrying oxidation and reductions of complex 1, a series of iron complexes
containing bis(amidinato)carbene ligands
spanning four oxidation states was obtained (Scheme 2). An extensive spectroscopic, magnetic,
and computational analysis of this series of complexes revealed that the bis(amidinato)-N-heterocyclic carbene complexes are exceptional
electron donating ligands, which results in behavior that differs significantly
compared to the bis(imino)pyridine complexes.
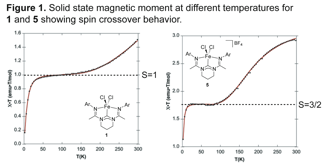
The magnetic properties of the iron(II)
and iron (III) complexes 1 and 5 illustrate the consequence of the electron donating capabilities of
the bis(amidinato)-N-heterocyclic
carbene ligands. Unlike the bis(amino)pyridine
complexes, which are high spin complexes at all temperatures greater than 10 K,
the bis(amidinato)-N-heterocyclic
carbene complexes demonstrate evidence for an intermediate spin state that pervades
at temperatures less than 150 K and undergoes a spin transition to high spin
complexes above room temperature (Figure 1). This behavior was further
corroborated with variable temperature X-ray crystallography and spin
unrestricted DFT calculations, and is consistent with larger ligand field splittings resulting from the more electron donating bis(amidinato)-N-heterocyclic carbene ligands.
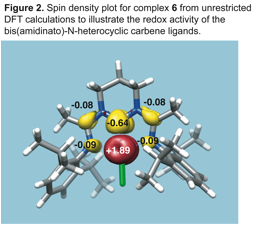
One electron reduction of 1 produced the formally iron(I) complex 6 (Scheme 2). As was the case for bis(amino)pyridine iron
complexes, spectroscopic evidence supported by spin unrestricted DFT
calculations revealed that bis(amidinato)-N-heterocyclic
carbene ligands are redox active in complex 6 (Figure 2). This finding
is notable because it is the first example of an isolated complex containing an
Arduengo-type N-heterocyclic carbene ligand that is
redox active. Interestingly, in contrast to the high oxidation state
complexes, which take advantage of the s-donating
capabilities of the bis(amidinato)-N-heterocyclic carbene
ligands, the low oxidation state complexes utilize the ability for the ligand
to act as a p-acceptor. By having the capacity to interact with
the metal center via many bonding modes, several oxidation states and spin
states of iron can be stabilized by these ancillary ligands.
With a firm understanding of their electronic structure, we
began to examine the catalytic performance of iron complexes containing bis(amidinato)-N-heterocyclic carbene ligands. Considering the superior
electron donating capabilities of the ligands, we originally hypothesized that they
would be suitable for stabilizing high oxidation states of iron. In preliminary studies, we revealed that
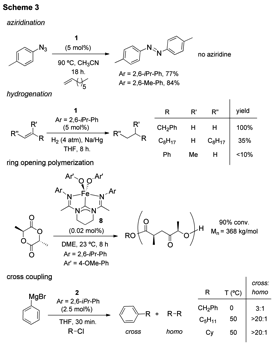
the iron complex 1 catalyzed the
formation of diazene products when exposed to azides in the presence of
olefins. This promising result
suggested that the bis(amidinato)-N-heterocyclic carbene
complexes could support high oxidation states of iron through the formation of
an iron amido intermediate. Ligand modifications to make the
complexes more sterically accessible for group transfer reactions made the
complexes more reactive for the formation of diazene products, but they unfortunately
did not result in any significant group transfer reactions.
In contrast to these findings, in situ reduction of complex 1 in the presence of hydrogen and
alkenes led to the rapid hydrogenation of alkenes. Although the reaction demonstrated
limited substrate scope, catalytic hydrogenation was promising considering that
the bis(dinitrogen)
iron complexes historically used for these reactions were not used. Future work in this direction will be dedicated
to synthesizing the bis(dinitrogen) iron complexes and applying them toward the
hydrogenation of alkenes and other reactions such as [2+2] cycloaddition of
alkenes and dienes. In a parallel program not supported by the PRF, our lab has
discovered that bis(imino)pyridine iron complexes are good catalysts for the
ring opening polymerization of cyclic diesters.{JACS2013, 135, 16553}Our initial studies revealed that
catalytic performance was superior when electron rich catalysts were utilized. We hypothesized that iron complexes
containing the exceptional s-donating bis(amidinato)-N-heterocyclic carbene ligands would be superior
catalysts for the polymerization of cyclic diesters. This hypothesis proved to be correct as
the iron alkoxide complexes 8 were
found to be very active for the polymerization of cyclic diesters
leading to the efficient production of high molecular weight polymer (>300
kg/mol) using small quantities of the iron catalyst (0.02 mol%).{Polyhedron2014, In Press, DOI:10.1016/j.poly.2014.07.002}These finding
demonstrate how the electron donating capabilities of the ligand can be
utilized to improve catalytic performance.
Considering our interest in ring opening polymerization, the versatility
of the bis(amidinato)-N-heterocyclic carbene ligands will be further
pursued in epoxide and lactone polymerization reactions. Finally, the bis(amidinato)-N-heterocyclic carbene and bis(imino)pyridine
ligands were evaluated as ancillary ligands in catalytic cross coupling
reactions. There have been many
mechanisms proposed that involve multiple oxidation states of iron for
iron-catalyzed cross coupling reactions.{ChemSusChem2009, 2, 396} We hypothesized that the redox activity
of the bis(amidinato)-N-heterocyclic
carbene and bis(imino)pyridine ligands would funnel
reactions towards one reaction pathway thereby prevent deleterious side
reactions resulting from multiple competing reaction pathways. Additionally, the redox active ligands
were expected to provide access to unusual oxidation states of iron that are
anticipated to alter catalytic performance. Moreover, the ligand motif is easily
modifiable for assymetric variants, and it is known
to discourage irreversible b-hydride
elimination reactions{Top. Organomet. Chem.2009, 26, 107}that have
historically been deleterious for cross coupling reactions. Despite these advantages, there have
been no reports that disclose the use of these ligands for catalytic cross
coupling reactions. Recently, we
discovered bis(imino)pyridine iron complexes can promote catalytic cross
coupling of phenyl Grignard with a variety of alkyl halides (Scheme 3). Reactions were efficient, and except for
benzyl chloride, were selective for cross coupling with little evidence for
alkyl halide homo-coupling or b-hydride
elimination. Moving forward, the catalytic
cross coupling reaction will be further optimized to limit the amount of
Grignard homocoupling product, and the versatility of
the bis(imino)pyridine complexes will be extended to
other cross coupling reactions, such as Suzuki cross coupling reactions.
 printer friendly
printer friendly