We investigated the use of NHC
catalysts to accomplish direct anodic oxidation of the Breslow
intermediate, absent redox mediators, and an expanded scope of alcohol coupling
partners. Although direct bulk electrolysis of Breslow
intermediates were previously unknown,
cyclic voltammetry studies of Breslow
intermediates derived from thiazoliumprecatalysts showed that these intermediates have much
lower oxidation potentials than their parent aldehydes (-0.9 V and > 2 V vs
SCE, respectively).
Initial
screenings revealed 1,8-diazabicyclo[5.4.0]undec-7-ene
(DBU) and CH3CN as the optimal base and solvent, respectively. An
extensive catalyst screen indicated that thiazolylidines
were more effective than their imidazolylidene, triazolylidene, and acyclic diaminocarbene
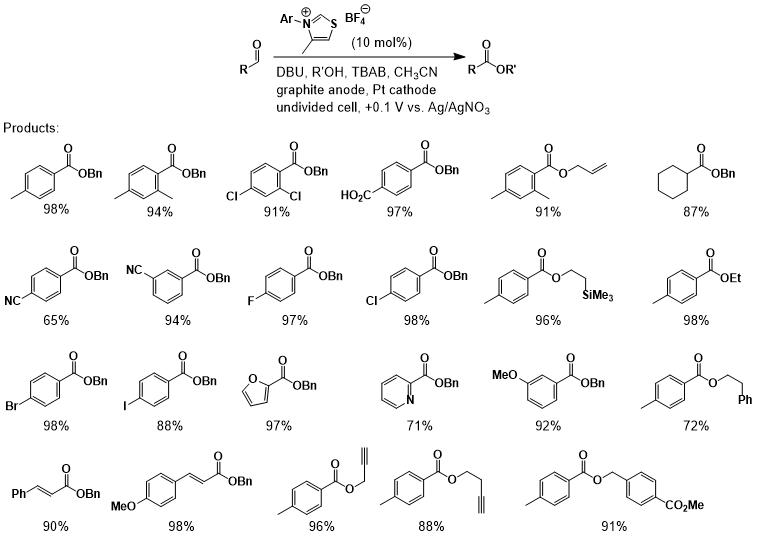
counterparts. In general, good to excellent yields were obtained for a wide
variety of aldehyde substrates (Figure 1), including substrates with ortho substitution and electrophilic groups, such as
carboxylic acid, cyano, and ester functionalities. A
notable limitation within the aldehyde scope is revealed when electron-rich benzaldehydes, such as 4-methoxy- and
4-dimethylaminobenzaldehyde, were employed. The reduced electrophilicity
of these substrates results in at best sluggish conversion to the desired
products, consistent with other approaches involving NHC-catalyzed oxidations
of electron-rich benzaldehydes. Reactivity was
restored by moving the electron-donating group out of conjugation with the
aldehyde (e.g., 3-methoxybenzaldehyde) providing the desired ester in 87%
isolated yield.
Moving away
from aromatic aldehydes, it was found that cyclohexanecarboxaldehyde
and α,β-unsaturated aldehydes reacted
smoothly without evidence of competitive side reactions, such as aldol
reactions, Stetter reactions, lactone formation, or conjugate reduction. A
broad range of alcohols were also found to participate in the oxidative
esterification. Most notably, functionalized and activated alcohols such as
benzyl alcohol, allyl alcohol, propargyl
alcohol, 4-pentyn-1-ol, and 2-(trimethylsilyl)ethanol proceeded smoothly. However, when s-BuOH and t-BuOH were
employed, little to no ester was obtained even at elevated temperatures.
Expanding
upon the scope of NHC-catalyzed anodic oxidation of aldehydes, we demonstrated
the direct conversion of aldehydes to thioesters. The
switch to thiol coupling partners in place of alcohols was nontrivial, as the
corresponding thiolates were found to be readily
oxidized by the azoliumprecatalysts.
Specifically, control experiments confirmed the formation of disulfides in the
presence of azolium salts and strong bases, and
disulfide formation was found to preclude thioester formation.
Figure 1.NHC-catalyzed anodic oxidation of
aldehydes to esters.
Following
an extensive base screen, it was found that the base strength and concentration
strongly influenced the product distribution. Optimal conditions involved using
50 mol % of 4-dimethylaminopyridine at slightly elevated temperatures. These
conditions ultimately furnished access to thioesters
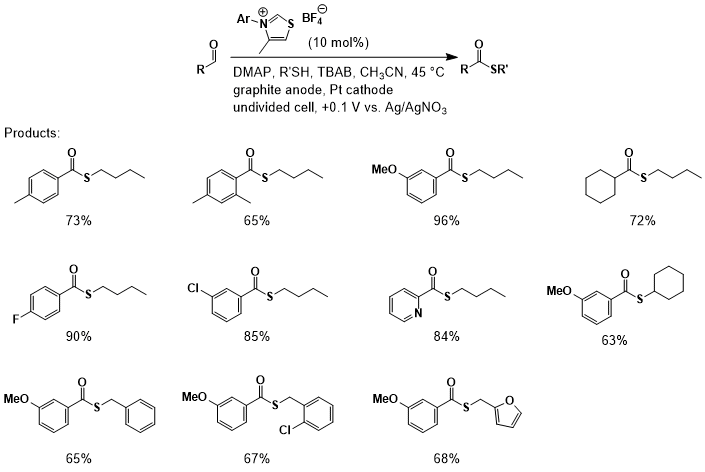
derived from a broad scope of aldehyde and thiol precursors, with yields
ranging from 56 to 89% (Figure 2). Unactivated and ortho-substituted aldehydes
proceeded smoothly, as did cyclohexane carboxaldehyde.
Electron-deficient aldehydes, including heteroaromatic
2-pyridinecarboxyaldehyde, provided good to excellent yields. Unfortunately,
the use of α,β-unsaturated aldehydes
resulted in poor yields presumably due to conjugate addition of the thiol. Thiols including primary thiols,
benzyl, 2-chlorobenzyl, and 2-furfuryl thiol each gave good yields of the
desired thioesters. Additionally, a secondary thiol, cyclohexanethiol, furnished the desired thioester in 63%
yield.
The
undesirable thiolate oxidation by thiazolium-based
NHC precursors that was observed during the development of our thioesterifications led us to investigate the reduction
potentials of azoliumprecatalysts.
Unwanted oxidation may be avoided in future studies through judicious selection
of azoliumprecatalysts
based upon their reduction potentials. We recently reported the reduction
potentials of various classes of azolium cations,
including a series of electronically varied thiazolium
species, and introduced the first report of N-aryl
thiazolinium salts.
Figure 2.Aldehyde and thiol substrate scope for
NHC-catalyzed anodic oxidations of aldehydes to thiosesters.
We
evaluated the redox properties of multiple azolium
salts using cyclic voltammetry. Each cyclic voltammogram (CV) was taken with a
sweep rate of 100 mV/s and ferrocene (0.010 M) was
added as an internal standard. Irreversible reduction peaks were observed in
the CVs of each azolium species. We investigated
several commonly used classes of azolium species for
direct comparison, and an expanded set of thiazolium
salts that demonstrated the extent to which electronic modification of the N-aryl moiety can be used to tune the
reduction potential of the thiazolium ring (Figure
3).
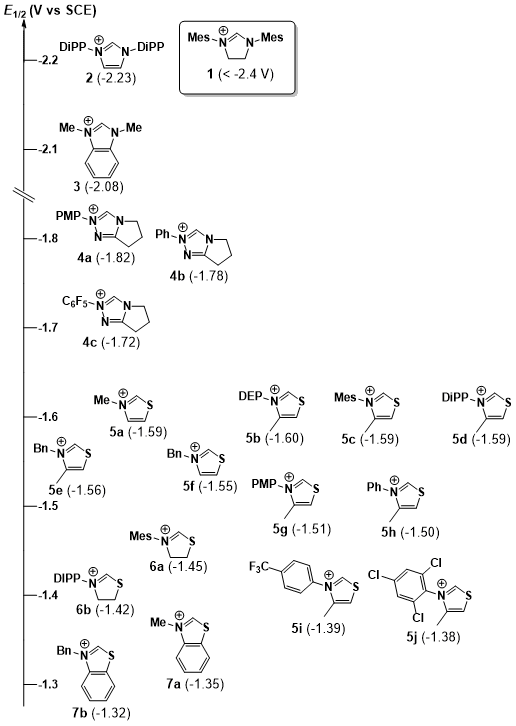
Figure 3. Half-wave reduction potentials of azolium
salts (counterions omitted for clarity). Values in
parentheses are E1/2
values (V vs SCE) for the corresponding cation. Mes =
2,4,6-trimethylphenyl, DiPP
= 2,6-diisopropylphenyl, PMP = p-methoxyphenyl, DEP = 2,6-diethylphenyl.
Within the
series of N-aryl thiazolium
salts (5a – j), a noticeable electronic influence was observed upon variation
of the N-aryl ring. The incorporation
of inductively electron donating groups (5b,
5c, 5d) shifts the E1/2 ca. 100 mV more negative. In contrast, electron
deficient groups (5i and 5j) shifted
the E1/2 ca. 100 mV more
positive. Somewhat surprisingly, incorporation of a p-methoxyphenyl group at nitrogen (5g) resulted in little change in E1/2 in comparison with the N-phenyl analogue (5h). This result may be due to the inability of the 5g system to adopt coplanarity
of the N-aryl and thiazolium
rings, due in part to the backbone methyl group of the latter. The > 200 mV
range of E1/2 values for
this set is nearly as broad as that observed across the span of benzothiazolium, thiazolinium,
and thiazolium salts combined. Interestingly, the N-aryl thiazolinium
salts were found to be easier to reduce than their unsaturated thiazolium counterparts (5c and 5d).This was surprising to us
considering the relative difficulty in reducing the corresponding saturated imidazolinium species 1.
In summary,
we have demonstrated the organocatalyzed anodic oxidation of aldehydes at low
controlled potentials. This method circumvents the use of stoichiometric
exogenous oxidants and high cell potentials, and produces minimal byproducts.
Relatively low catalyst loadings were successful for oxidative esterification
and thioesterification of aldehydes, with H2
gas as the only stoichiometric byproduct. In addition, the redox properties of
a broad series of azolium salts were systematically
investigated via cyclic voltammetry. These electrochemical studies may aid in
the development of azolium-based materials, NHC precatalysts, electrosynthesis of NHCs, and correlation of
electrochemical properties and other fundamental reactivities.
 printer friendly
printer friendly