Reports: DNI152447-DNI1: Metal Catalyzed C-H Bond Activation and Allyl Addition: Development of New Transition Metal Catalysis for Practical and Uniquely Efficient Carbon-Carbon Bond Forming Reactions
Simon J. Meek, PhD, University of North Carolina (Chapel Hill)
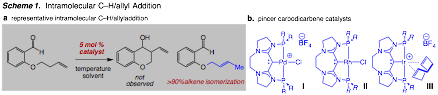
We continue to investigate the development of new catalysts for the conversion of alkenes into allyl nucleophiles through C–H bond activation.Our initial design was based on the ability of palladium complexes to initiate allylic C–H activation as well as promote the addition of allyl fragments to C=O bonds.To this end, we successfully synthesized and developed a new class of pincer carbodicarbene (CDC) (carbon(0) donor) ligand scaffolds; such ligands exhibit distinct architectural properties amenable to steric and electronic modification.Well-defined pincer carbodicarbene (CDC) palladium complexes were previously synthesized but proved ineffective towards a C–H / allylation process despite being able to catalyze the individual steps of (i) allyl group transfer, and (ii) alkene isomerization.We turned our attention to other transition metals known to facilitate C–H activation and carbonyl allyl addition, specifically, Rh, and Ir.(CDC)-pincer complexes were successfully synthesized and evaluated (Scheme 1).Both rhodium and iridium afford rapid alkene isomerization but do not form the desired carbon-carbon bond.In order to maximize the success of a new C–H/allylation process, we elected to study intramolecular variants (Scheme 1).To date, in the presence of a number of transition metals and ligands, which include the CDCs previously developed in this program, no desired reaction was observed.However, in most cases with pincer CDC-based catalysts (e.g., complexes I, II, and III) rapid alkene isomerization occurred (>90%, Scheme 1).
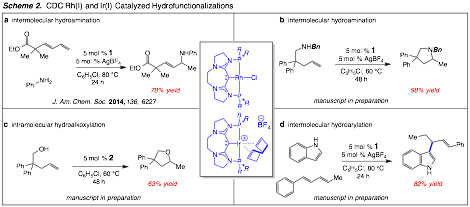
Despite extensive investigations into a number of transition-metals catalysts, we have not been able to thus far initiate a C–H/allylation process.This has led us to take a step back and make certain we have a handle on all the experimental variables as well as understanding the reactivity of the novel pincer CDC ligands developed in these studies.While pincer CDC catalysts have so far not yielded the desired C–C bond formation they are effective at isomerizing alkenes, most likely through a desiredh1-allyl fragment.To this end we have explored the basic reactivity of the CDC ligands and pincer complexes in Scheme 1 towards understanding their fundamental reactivity. We have discovered that Rh and Ir complexes II and III, are effective at catalyzing the addition of N–H, O–H, and C–H bonds to unsaturated C–C double bonds (Scheme 2).Rhodium(I) complex II, proves to be the most versatile catalyst as it initiates both intra- and intermolecular hydroaminations, as well as, intermolecular hydroarylation.Treatment of rhodium(I) chloride complex II with equimolar AgBF4 effectively initiates the hydrofunctionalization processes.Of note the reactions are tolerant of aryl and alkyl amines; unactivated terminal alkenes participate but only for intramolecular variants.In all cases the rhodium(I) center is made cationic through halide abstraction to facilitate alkene binding and subsequent nucleophile attack at the activated alkene.Initial mechanism studies indicate that an N–H insertion process is not operational.By analogy, Ir(I)–alkene complex III was found to catalyze intramolecularhydroalkoxylation to deliver tetrahydrofurans in good yield.The reaction mechanism is unknown but could proceed via O–H insertion, which would be important towards developing a C–H/allylation reaction.Concurrent with our investigations towards understanding the reactivity of CDC catalysts, we also have garnered information about carbon(0) electron donor capabilities through CO stretching frequency studies. In comparison to other reported cationic Rh(I) complexes our data revealed the pincer CDC ligands to be stronger electron donors.The results of the intermolecular hydroamination, X-ray structures, and electronic donor properties of the CDC-Rh(I) complexes were recently communicated in Journal of the American Chemical Society.Manuscripts are in preparation to disclose the other three reactions classes in Scheme 2.
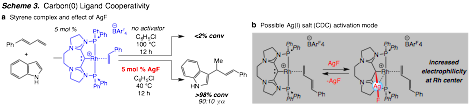
To achieve a catalyst capable of C–H/allylation we have explored potential metal–ligand cooperativity made available through a carbon(0) donor group in CDCs. Mechanism studies regarding hydroarylation have revealed activation of (CDC)-Rh(I) complexes occurs through binding of the carbon(0) donor to a silver(I) salt.The resulting hetero-bimetallic system, most likely through an interaction between Ag(I) and Rh(I), leads to a more reactive catalyst (Scheme 3).We are currently trying to observe the Ag(I) binding interaction as well as understand and utilize the activation strategy made available by this discovery.
The research conducted with the support of the PRF during this funding period has allowed us to determine that pincer CDC ligands are effective in catalyzing hydrofunctionalization processes of C–C double bonds. Furthermore, mechanistic understanding of CDC ligand electronic properties have led to the discovery of metal–ligand cooperativity unique to carbon(0)-based catalysts.This cooperativity provides an avenue for achieving new reactivity.We anticipate that the progress reported herein will provide more efficient ways to effectively functionalize unsaturated hydrocarbon feedstocks, by utilizing new strategies in catalysis design. Our continuing efforts in pursuing a C–H activation/allylation process through CDC pincer complexes remains one of the foundations of our group's research effort.
 printer friendly
printer friendly