Reports: UNI152943-UNI1: Foldameric Catalysts for Enantioselective Trifluoromethylations of Ketones and Enones
 printer friendly
printer friendlyOur efforts to develop such catalysts commenced with the synthesis and screening of small model peptoids 1 and 2 containing amine N-oxides as the nucleophilically catalytic moieties (Figure 1A). A number of oxidation methodologies for constructing these models were assayed, and a procedure utilizing bicarbonate-activated peroxide ("BAP") was found to give quantitative yields (Figure 1B).3 However, catalysts 1 and 2 were found to be completely inactive as nucleophilic trifluoromethylation catalysts under all conditions tested, while control experiments utilizing N,N-dimethylbenzylamine oxide demonstrated that our reaction conditions were viable.4,5 We thus proceeded to optimize the trifluoromethylation reaction conditions in the hope that conditions would be found in which 1 and 2 were active. During this process, we discovered that acetonitrile is a highly effective solvent for the reaction – an important development given that peptoid secondary structure is not well elucidated in any of the other polar solvents known to facilitate nucleophilic trifluoromethylation. Even in acetonitrile, however, catalysts 1 and 2 were found to be inactive, suggesting that there was too much steric congestion near the active site for effective catalysis.
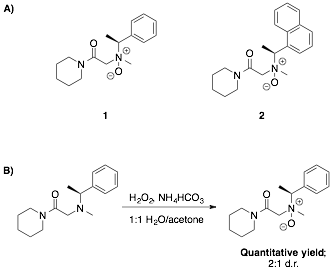
Figure 1. A) Model peptoid catalysts. B) Oxidation of peptoids to generate amine N-oxide catalysts.
We thus adjusted the design of our catalytic residue by replacing the directly attached chiral moiety with a methyl group, and incorporated the residue into N-‡-chiral aromatic peptoids known to adopt secondary structures in acetonitrile (Figure 2). These catalysts were sufficiently pure upon cleavage from resin to conduct initial tests of catalyst enantioselectivity in the nucleophilic trifluoromethylation of benzaldehyde as "proof of principle" experiments. We were gratified to observe that, while the ee observed for dimer 3 was quite low, an ee of 36% was obtained using trimeric catalyst 4. Taken together, these results suggest that helical secondary structure (Figure 3), rather than a nearby chiral center in the primary structure, will dictate the enantioselectivities of this catalyst class.
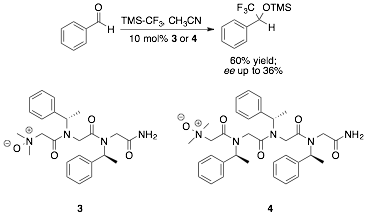
Figure 2. Initial tests of catalyst enantioselectivity in the nucleophilic trifluoromethylation of benzaldehyde.
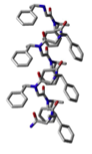
Figure 3. Helical structure adopted by N-‡-chiral aromatic peptoids (gray = carbon, blue = nitrogen, red = oxygen; hydrogens omitted for clarity).
In the next phase of my research program, I plan to have my students finalize a peptoid oxidation protocol based on what has been learned about the side reactions encountered during mCPBAoxidations. We will then construct longer N-‡-chiral aromatic peptoid oligomers (i.e. tetramers, pentamers, and hexamers) and determine how increasing length correlates with catalyst reactivity and enantioselectivity. We also plan to incorporate additional side chain types, especially those containing hydrogen bond donors, in an effort to engender bifunctional catalysis that could dramatically enhance catalyst performance.
The support provided by this grant has allowed me to lay the foundations for a successful research program that is already yielding publishable data, and that is critical to establishing myself in the field of organocatalysis. Moreover, the program has excited and inspired undergraduates to join me in pursuing this research, which in turn has inspired several of them to pursue chemistry degrees and careers in the sciences. Going forward, I envision applying the synthetic methodologies we are developing towards the synthesis of large libraries of peptoid catalysts useful for not only trifluoromethylations, but also for other classes of reactions that are difficult to conduct using typical small-molecule catalysts. These pursuits could ultimately open avenues toward new and efficient syntheses of petroleum-derived products.
(1) Zuckermann, R. N.; Kerr, J. M.; Kent, S. B. H.; Moos, W. H. J. Am. Chem. Soc. 1992, 114, 10646.
(2) Maayan, G.; Ward, M. D.; Kirshenbaum, K. Proc. Nat. Acad. Sci. U.S.A. 2009, 106, 13679.
(3) Balagam, B.; Richardson, D. E. Inorg. Chem.
(4) Surya Prakash, G. K.; Mandal, M.; Panja, C.; Mathew, T.; Olah, G. Journal of Fluorine Chemistry 2003, 123, 61.
(5) Surya Prakash, G. K.; Panja, C.; Vaghoo, H.; Surampudi, V.; Kultyshev, R.; Mandal, M.; Rasul, G.; Mathew, T.; Olah, G. A. J. Org. Chem. 2006, 71, 6806.










