Reports: UR551765-UR5: Atomistic Simulations of Small Molecules' Behavior Within Al Substituted Zeolites
 printer friendly
printer friendlyTo study cation siting within LTA-4A we started by using available potentials(1) to reproduce absorption isotherms. The next step, was the attempt to reproduce experimentally determined sites for cations in the absence of adsorbed gases. To our surprise using this yielded inaccurate cationic sites. Figure 1 below, shows the experimental positions (in blue) of a cation blocking the passage of gas molecules. The same figure shows the simulations results (in red) – as it is apparent in the figure the sites do not coincide rendering Garcia-Sanchez's force field as not one that we can use further.
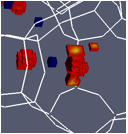
Figure 1. Na+ experimental (blue) and simulation (red) siting positions within an 8MR of LTA-4A do not coincide rendering Garcia-Sanchez's(1)force field unsuitable for our calculations.
Fortunately, last year Fang and collaborators published a new force field that seemed promising.(2)Using their potentials we were able to get much more accurate siting of cations within LTA-4A; however, one of the sites was able to accommodate more than one cation (which is not the case experimentally); see figure 2, below. We are in current communication with Sholl's group and they are in the process of revising their force field. (Our results highlighted a shortcoming of their work that they are eager to overcome). We are planning to resume its study as soon as we are able to.
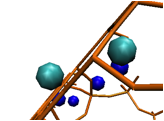
Figure 2. Views of an LTA-4A 8MR that is occupied by two Na+ (light blue). Other cations are shown in dark blue. This unrealistic siting renders Fang's(2)force field in its current form unsuitable for our calculations.
In addition, this past year we switched from an in-house software package to one available upon request from Snurr's group in Northwestern University. RASPA, this new code, is slower than ours, but the burden of development is now mostly on their shoulders. Furthermore, RASPA promises to be a very versatile tool for us. When it was clear that we did not yet had a force field suitable to studying siting within LTA-4A and MFI, we switched our focus to other materials. In the past, this would have necessitate months of an undergraduate's work, but RASPA allowed us to start investigating materials we were not familiar with within a couple of days.
RHO is a promising material that has been found to exhibit "cation gating(3-5)". It has been hypothesized that gating occurs because the "gating" cation sits in a position that blocks diffusion of gas molecules, but it moves in the presence of some gases (most notably CO2) allowing the molecules to diffuse from cage to cage. Experiments can only look at average positions of cations, which mask this type of behavior. With simulations, however, we were able to obtain distributions that, to our knowledge, constitute the first quantitative evidence for cation gating in this zeolite (shown in figure 3, below).
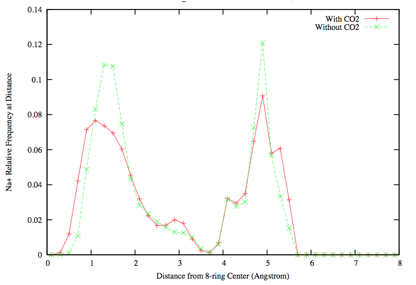
Figure 3. Distance of cations from blocking position in the center of the ring. The change in distributions in the presence of Carbon Dioxide is an indication of gating
Two students were supported in the summer of 2014 by this grant. This last year our work has resulted in two students' talks at the regional MU3C meeting, two students' talks to the members of the Carleton chemistry department doing research over the summer, and one poster presented at an all-science poster session at Carleton College. I also anticipate that we will be submitting a manuscript summarizing our RHO results and presenting that work at the Porous Materials Workshop (CPM-7) and at the Foundations of Molecular Modeling and Simulations Conference within a year.
1. A. Garcia-Sanchez et al., The Journal of Physical Chemistry C 113, 8814-8820 (2009).
2. H. J. Fang et al., Physical Chemistry Chemical Physics 15, 12882-12894 (2013).
3. M. M. Lozinska et al., Journal of the American Chemical Society 134, 17628-17642 (Oct 24, 2012).
4. M. M. Lozinska et al., Chemistry of Materials 26, 2052-2061 (Mar 25, 2014).
5. M. Palomino, A. Corma, J. L. Jorda, F. Rey, S. Valencia, Chemical Communications 48, 215-217 (2012).










