Reports: ND752426-ND7: Thermodynamics of Nanoconfined Equilibrium Polymerizations
 printer friendly
printer friendlyThe test of this hypothesis has focused on making measurements for methyl methacrylate polymerization under nanoconfinement. Bulk poly(methyl methacrylate) has a ceiling temperature of approximately 220 °C, as shown in Figure 1. Under nanoconfinement in 13 nm pores of controlled pore glass (CPG), the ceiling temperature shifts to lower temperatures, as also shown in Figure 1. The shift in the ceiling temperature to lower temperatures is consistent with a decrease in the entropy of propagation under nanoconfinement. The fit to the nanoconfined data shown assumes that to a first approximation, the entropy change (which is as high as 15 J/K/mol) is exponential with decreasing temperature; this fits the data better than a constant entropy change and is a reasonable assumption since kinetic chain length follows a similar temperature dependence.
In addition to the nanoconfined equilibrium polymerization reaction, as part of the project, we also investigated the Diels-Alder dimerization reaction of cyclopentadiene (CPD) to form dicyclopentadiene (DCPD). Only one calorimetric study had previously been performed for the neat system over a broad temperature range, and in that work, Flammersheim and Opfermann [5] performed dynamic DSC scans at different heating rates to temperatures as high as 165°C and fit their data with an equilibrium kinetic model. Our original goal was to confirm the reaction kinetics and thermodynamics of the bulk reaction and then to investigate the reaction under nanoconfinement; however, as is briefly described below, we found that the reaction was, in fact, not an equilibrium reaction in the liquid phase, and hence further experiments under nanoconfinement were not carried out.
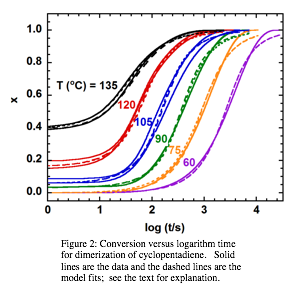
Both isothermal and dynamic DSC experiments were performed. Figure 2 shows the results of the isothermal experiments, plotted as conversion as a function of logarithmic time for several reaction temperatures. The data are fit to a second-order autocatalytic irreversible kinetic model, neglecting and incorporating the effect of pressure in the DSC pan used to study the reaction (which could withstand up to 24 bar pressure). The short dashed line represents the model without pressure effect and the long dashed line represents the model incorporating the effect of pressure; the latter fits the data significantly better. Although the Diels-Alder cyclopentadiene dimerization has been considered to be an equilibrium reaction in several works, [5,6-8] we find that for our temperature range, predominantly below 180 °C, goes essentially to completion based on the DSC, GC-MS, and NMR results. The seeming inconsistency with literature conclusions can be explained by looking more closely at the rate constant of the reverse reaction, which is reported in the literature [5,7] to be ten thousand times slower than that of the forward reaction even at high reaction temperatures, consistent with our findings.
Figure 1: Enthalpy and conversion versus temperature for bulk and nanoconfined MMA polymerization. |
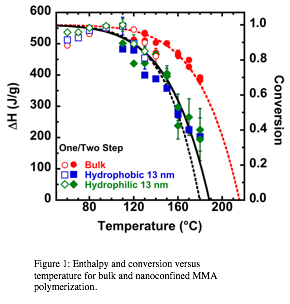
Figure 2: Conversion versus logarithm time for dimerization of cyclopentadiene. Solid lines are the data and the dashed lines are the model fits; see the text for explanation. |
1. C. Y. Kong and M. Muthukumar, "Polymer translocation through a nanopore. II. Excluded volume effect," J. Chemical Physics, 120, 3460 - 3466 (2004).
2. T. Ishinabe, "Conformational Properties of a Polymer Chain Confined between Two Plates," Journal of Chemical Physics, 83 (1), 423 - 427 (1985).
A. Cacciuto and E. Luijten, "Self-Avoiding Flexible Polymers under Spherical Confinement," Nano Letters, 6 (5), 901 - 905 (2006).
3. S. M. Bezrukov, I. Vodyanoy, R. A. Brutyan, and J. J. Kasianowicz, "Dynamics and Free Energy of Polymers Partitioning into a Nanoscale Pore," Macromolecules, 29, 8517 - 8522 (1996).
4. H. J. Flammersheim, J. Opfermann, The Dimerization of Cyclopentadiene ---- A Test Reaction for the Kinetic Analysis of DSC Measurements and The Performance of a Kinetic Evaluation Program, Thermochim. Acta 337, 149-153 (1999).
5. T.G. Lenz, J.D. Vaughan, Employing Force-Field Calculations to Predict Equilibrium Constants and Other Thermodynamic Properties for the Dimerization of 1,3-Cyclopentadiene, J. Phys. Chem. 93, 1592-1596 (1989).
6. I. Palmova, J. Kosek, J. Schongut, M. Marek, K. Stepanek, Experimental and modeling of oligomerization and copolymerization of dicyclopentadiene, Chem. Eng. Sci. 56, 927-935 (2001).
7. Z. Belohlav, P. Zamostny, P. Fulin, K. Stepanek, Kinetic Model for Dicyclopentadiene Monomeration Process, Chem. Biochem. Eng. Q. 14, 29-33 (2000).










