Reports: ND151929-ND1: Self-Assembly of Catalysts and Ligands by Reversible Covalent Interactions of Organoboron Compounds
Mark S. Taylor, PhD, University of Toronto
Progress Report
Introduction. Methods
for the rapid synthesis of structurally diverse ligands or catalysts can significantly
accelerate the pace of reaction discovery and optimization. A powerful approach
involves the self-assembly of candidate structures through noncovalent
or reversible covalent interactions such as metal–ligand coordination, hydrogen
bonding and ion pairing. Catalysts are generated by simple
pairwise mixing of components, thus obviating the need for time-consuming
purification and isolation steps. This project explores applications of
reversible covalent interactions between boronic
acids and diols as the basis for catalyst assembly. Organoboron–diol
interactions are specific, strong and reversible, and have been exploited
extensively in the molecular recognition field. The tolerance of these
reversible covalent interactions to a range of solvents and reaction conditions,
and the availability of diverse, chiral diolfeedstocks constitute unique potential advantages in the
context of applications in catalyst self-assembly.
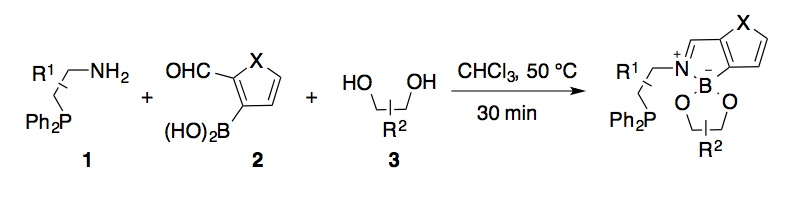
Libraries of chiral phosphines prepared by three-component condensations of
2-formylphenylboronic acid. Self-assembly of
chiral phosphine ligands through boronic acid–diol interactions was among the initial aims of the
project. The non-trivial synthesis of boronic
acid-functionalized phosphine components presented an early hurdle. To solve
this problem, we adopted a modified strategy, using three-component
condensations of phosphine-functionalized amines, diols
and 2-formylphenylboronic acid, generating
imine–boronate ligands where the boronic acid component serves as a linchpin. The ligand
assembly reactions proceed rapidly and to very high conversion, generating
water as the only byproduct. NMR spectroscopy studies and computational
modeling of transition metal complexes, along with preliminary
structure–activity relationships for a representative Pd(0)-catalyzed allylic alkylation reaction, suggested a P,N-coordination
mode for these novel ligands.
Scheme 1. Self-assembly of functionalized phosphine ligands by imine-boronate condensation.
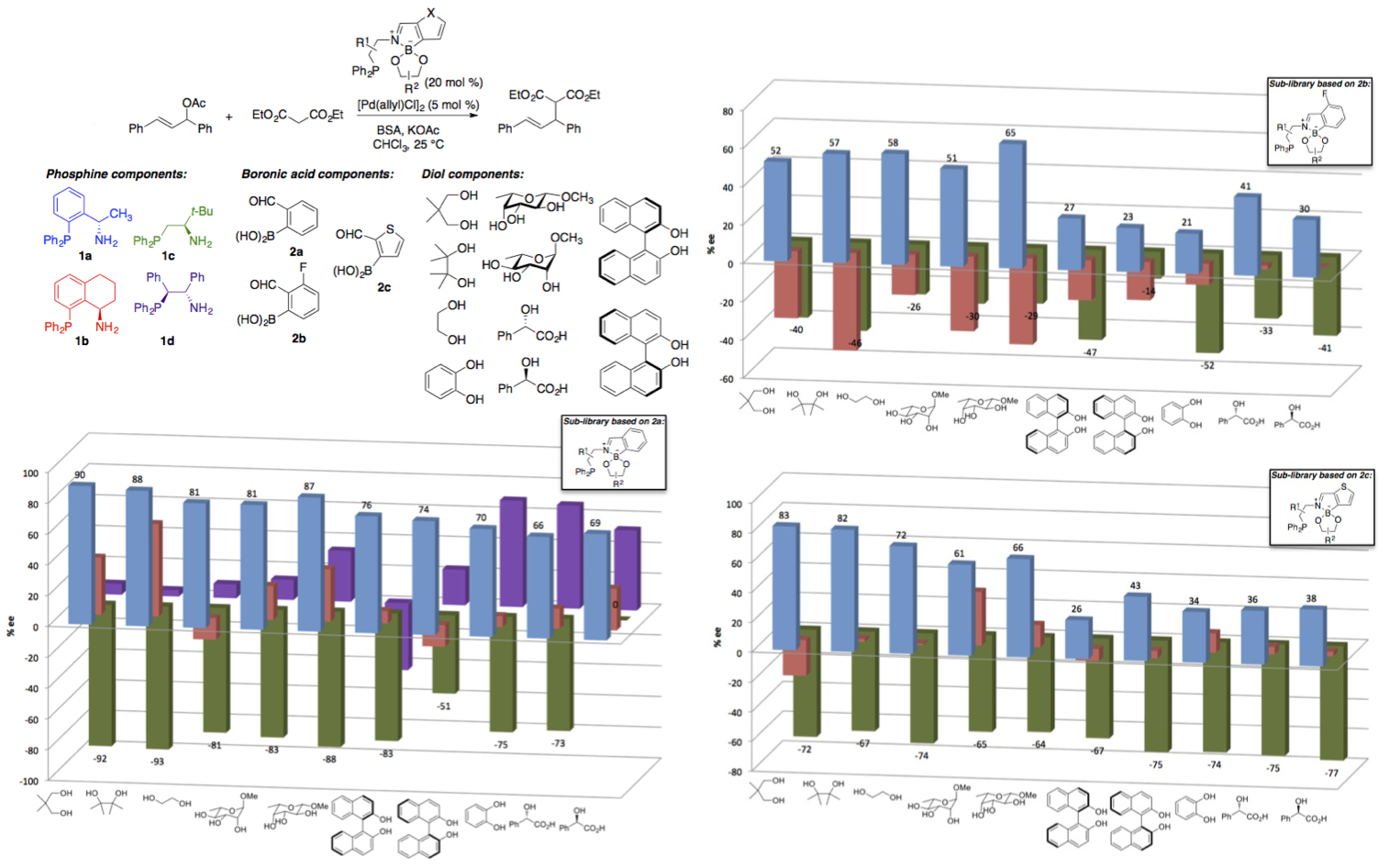
A
library of 100 phosphines was generated from selected
combinations of four aminophosphines, three formyl-functionalized boronic
acids and ten diols. The ligands were tested in a Pd(0)-catalyzed
allylic substitution reaction (Scheme 2). Each of the
three components of the ligand was found to influence enantioselectivity,
and matching/mismatching effects were evident for certain pairs of components. A
ligand that gave rise to high enantiomeric excess
(93% ee) for this benchmark substrate combination was
identified.
Scheme 2.
Evaluation of a library of 100 self-assembled P,N-ligands for asymmetric allylic alkylation.
Our
recent research has been aimed at applying these libraries of structurally
novel self-assembled ligands to more challenging substrate combinations and/or types
of reactivity. From this perspective, one of the key limitations of our initial
library was the lack of electronic or steric diversity at the diarylphosphino moiety in building blocks 1a–1d. It is known that variation of the phosphine component of P,N-ligands can dramatically influence the reactivity and
selectivity of their derived transition metal complexes. We were particularly
interested in modifying the aryl groups of aminophosphines
such as 1c, which provided the
highest selectivities in our initial library screen. A
further advantage of components such as 1c
is their synthetic accessibility from amino acids, thus allowing straightforward
variation of the substituent on the amine-bearing chirality center. However, our
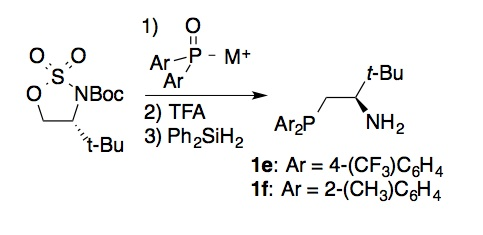
attempts to adapt the reported synthesis of 1c (via opening of a cyclic sulfamidate with
potassium diphenylphosphide) to other diarylphosphide nucleophiles were unsuccessful. To solve
this problem, we developed a new variant of this method in which a metalated secondary phosphine oxide was employed as the
nucleophile, and then reduced to the corresponding phosphine in the final step
of the process. This protocol enabled incorporation of electron-deficient (e.g., 1e) and sterically encumbered (e.g., 1f) diarylphosphino moieties that had
previously proved challenging. We anticipate that this modified route will
allow us to systematically vary the properties of the phosphine group while
constructing ligand libraries.
Scheme 3. Modified
synthesis of amino alcohol-derived diarylphosphines.
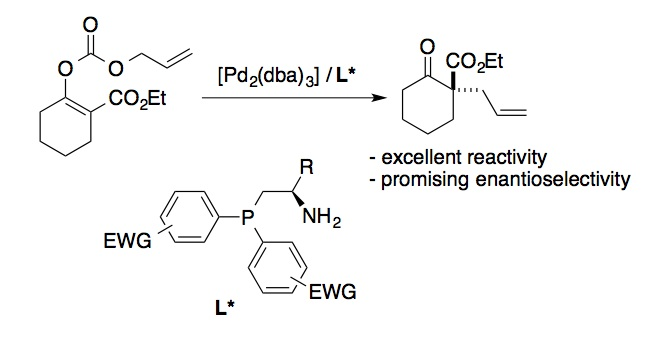
Among
the reactions for which imine–boronate ligands
provided interesting preliminary results was the enantioselective,
decarboxylativeallylation shown
in Scheme 4. However, control experiments revealed that it was the
amine-phosphine component of the ligand assembly (of general structure L*) alone that gave rise to the
observed enantioselectivity. While it is clear that
the ligand self-assembly strategy was not fruitful in this case, it is
noteworthy that, for this particular substrate, a structurally simple and
readily accessible ligand provided superior results to the optimum phosphinooxazoline ligand employed by Stoltz
and co-workers for such transformations. We are exploring straightforward
structural modifications of this type of amine-phosphine ligand to optimize its
activity and enantioselectivity in decarboxylativeallylation reactions.
Scheme 4. Enantioselective,
Pd-catalyzed decarboxylativeallylation using a chiral aminophosphine
ligand.
 printer friendly
printer friendly