Reports: ND153146-ND1: New Approaches to Alkene Functionalization via Dehydrogenative Metallation Catalyzed by First-Row Transition Metals
 printer friendly
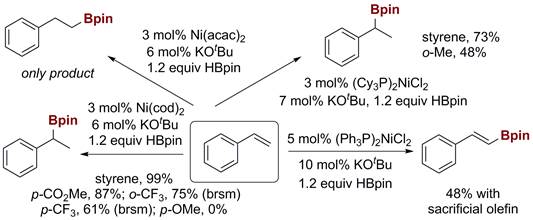
printer friendlyScheme 1. Divergent reactivity of Ni via the ligand choice.
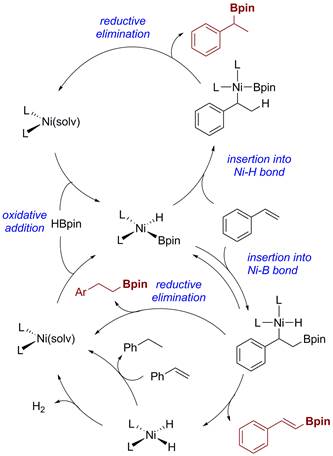
The proposed mechanistic pathways for all three products are illustrated in Scheme 2. Studies are ongoing to improve the scope, yields and tunability of Ni-catalyzed styrene functionalization. The information obtained from these investigations used as design principles for exploring similar reactivity with Cu and Fe catalysts in the context of dehydrogenative borylation.
Scheme 2. Mechanistic pathways for Ni-catalyzed transformations of styrenes.
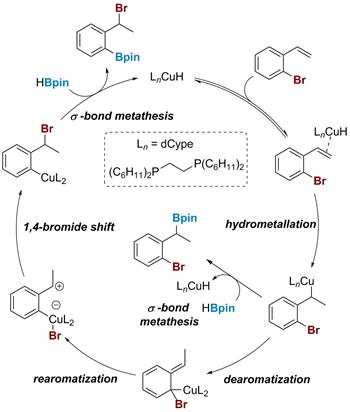
Scheme 3. Computational studies on the Cu-catalyzed 1,3-migration of 2-bromostyrenes.
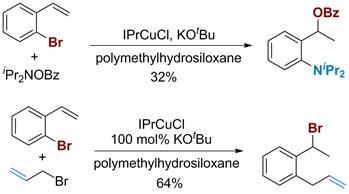
Scheme 4. Asymmetric Cu-catalyzed 1,3-migration of 2-bromostyrenes.
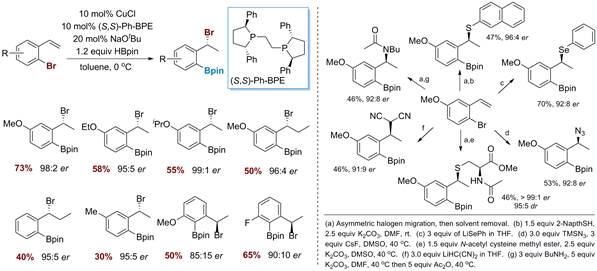
Scheme 5. Further functionalization of ArCu(I) species with electrophiles.