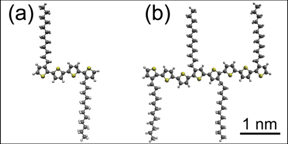Reports: DNI652732-DNI6: Atomic-Scale Visualization of Excitonic States in Individual Polymer Molecules
George Nazin, PhD, University of Oregon
Polymer
materials owe their success to the enormous amount of research carried out to
understand their properties. Despite this large body of knowledge, a
fundamental aspect of polymer properties--the relationship between the exact
polymer chain structure and electronic properties--remains poorly understood,
owing to the lack of tools capable of probing this relationship. In the
traditional picture of polymer photophysics, structural defects in polymer chains
play a decisive role in shaping the properties of polymers: conformational as
well as chemical defects can break electronic conjugation, and confine
electronic excitations over small chain segments, which act as individual
chromophores with electron energies determined by the extent of electronic
conjugation (Figure 1).
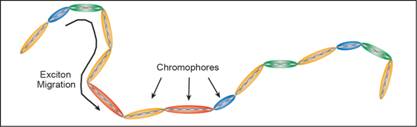
Figure 1.Segmentation of a conjugated polymer chain into a collection of
chromophores. Chromophore colors are determined by the chromophore lengths.
The
goal of our research program is to investigate the impact of different types of
structural disorder on the electronic properties of polymers using advanced
real-space spectroscopic techniques based on Scanning Tunneling Microscopy.
Visualization of electronic states in individual polymer molecules will provide
direct answers to a number of fundamental questions that so far could be
addressed either indirectly, or through theoretical simulations. In the first
year of the project, we studied the conformationally-dependent electronic
properties of model oligothiophene molecules.
The
experiments were performed using a home-built cryogenic ultra-high vacuum (UHV)
scanning tunneling microscope (STM). In two sets of experiments, two different
types of didodecyl-quaterthiophene oligomers (DDQT-1 and DDQT-2, Figure 2 (a)
and (b)) molecules were deposited on an atomically flat Au(111) surface, in
situ via sublimation. Each oligomer contained a conjugated backbone
composed of thiophene rings, and alkyl chains attached to the backbone.
DDQT-1, containing four thiophene rings, was used as a model useful for
isolating the effects of short-range disorder on the electronic structure,
while DDQT-2, containing eight thiophene rings, served as a basic model for
long polymer chains. In our experiments, alkyl ligands serve as direct markers
of the torsional conformation of individual oligothiophene molecules.
Figure 2.Models of
didodecyl-quaterthiophene oligomers (a) DDQT-1 and (b) DDQT-2. Hydrogen (light
gray), sulfur (yellow), carbon (dark gray).
STM
imaging of a sub-monolayer of DDQT-1 molecules on Au(111) revealed a variety of
conformations (Figure 3). Two types of the apparent conformations, straight-
and curved-back (SB and CB, Figure 3(a) and (b)), with both alkyl chains
oriented in the same direction, were found predominately in dimers and clusters
of dimers. The third conformation type (TB (twisted back), Figure 3(c)),
primarily found in crystal packing structure-like “ribbons” (Figure 3 (d)),
corresponds to the molecular structure with alkyl chains pointing in opposite
directions indicating that the rings of the thiophene backbone are twisted 180° with respect to each other as in
Figure 2(a). All three conformations correspond to different thiophene backbone
shapes enabling a study of the effects of three different conformations on the
oligothiophene electronic structure. Theoretical calculations indicate that
despite curving of the polymer backbone, and torsional disorder (twisting of
thiophene rings with respect to each other), individual thiophene rings can
still be coupled to each other, thus forming delocalized electronic states with
electronic coherence extended over several thiophene units.
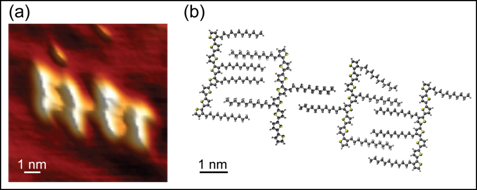
Figure 3. STM topographies of
DDQT-1 on Au(111) substrate revealed (a) straight- and (b) curved-back dimers,
and (c) twisted-back monomers, indicative of the crystal-packing conformation
found in (d) "ribbons". (e)-(h) are proposed models for (a)-(d),
respectively. All data were collected at 24 K.
To
test this prediction, we used Scanning Tunneling Spectroscopy (STS) to measure
the electronic density of states (DOS) for individual oligothiophene molecules.
The energies of the molecular orbitals for all quaterthiophene conformations
lie in the range of -0.9 to -1.1 eV (HOMO), and 2.1V-2.3 eV (LUMO), in good
agreement with the expected band-gap of quaterthiophenes at ~3 eV. However,
reproducible differences are found between the energies corresponding to three
different conformations. Calculations of these conformations agree with
observations, finding energy differences of up to 0.25 eV between the
conformations, as well as delocalized HOMO and LUMO orbitals. By spatially
mapping the energy-dependent DOS using STS, we found
that both the LUMO and HOMO of all studied DDQT-1 conformations are
indeed delocalized over each backbone. The differences in the orbital energies
found for the three conformations are thus attributed to the varied effective
strength of electronic coupling between the different thiophene rings for the
three conformations. A manuscript describing these results is currently in
preparation.
While
the DDQT-1 molecule represents a convenient elementary model for studying
short-range structural disorder, it does not capture the effects of
longer-range conformational disorder and the statistical effects of random
torsional disorder. Further, the orbital energies for DDQT-1 are relatively
high, which limits the number of DDQT-1 orbitals that can be probed in STS
measurements without disturbing the molecules. To investigate the effects of
long-range disorder on the oligothiophene electronic structure, we have carried
out experiments on the longer oligothiophene, DDQT-2. STM imaging of DDQT-2
deposited on a Au(111) surface shows self-assembled chains of molecules
tethered to each other with alkyl chains (Figure 4), analogously to the
crystal-like structures observed for DDQT-1. A wide variation of straight and
curved conformations was found. DOS mapping for these molecules shows
particle-in-a-box-like
states (from HOMO to LUMO+2), typically delocalized over the conjugated
backbones. Work on systematic STM/STS characterization of individual DDQT-2
molecules, coupled with theoretical simulations, is currently under way.
Figure 4. (a) STM topography of
DDQT-2 chain on Au(111) and (b) proposed molecular model. Hydrogen (light
gray), sulfur (yellow), carbon (dark gray).
The work described
above was performed by two graduate students (Christian Gervasi and Dmitry
Kislitsyn), a postdoctoral researcher (Yinghao Liu, now at Seagate), and an
assistant professor (George Nazin). Four graduate students (Christian Gervasi,
Dmitry Kislitsyn, Ben Taber and Jon Mills) and two undergraduate students
(William Crowley and Ka Hung Lee) will continue to work on the project and
investigate in further detail the conformationally-dependent electronic
properties of DDQT-2 molecules by using a combination STS measurements and
electronic structure calculations. Figure 5. Nazin group, Winter
2014.
 printer friendly
printer friendly