Reports: DNI653482-DNI6: Signature of Molecular Environment in Spectroscopy Measurement of Water and Aqueous Solutions Studied by Advanced ab Initio Methods
 printer friendly
printer friendlyA large part of the inaccuracy of current AIMD originates
from the DFT functional approximation of GGA. Several studies showed that GGA
is not good enough for H-bond structure predictions. On one hand GGA inherits
the self-interaction errors from (semi)local exchange correlation
approximations, which results in the over-structured pair correlation function
from excessive proton delocalization and artificial red shifts of the OH
stretching frequency. The self-interaction error can be largely alleviated by the
hybrid-XC, e.g. PBE0, which mixes a fraction of Hartree-Fock exact exchange. It
was found that PBE0 improves greatly the vibrational spectrum of liquid water,
which softs the H-bond strength and reduces the discrepancy between experiment
and theory for the first peak of OO and OH pair correlation functions. On the
other hand, both hybrid-XC and GGA miss the attractive vdW interactions
originating from the dynamic correlations of the electron density. Under the
vdW forces, the density of non H-bonded molecules will be increased in the
first coordination shell giving a further correction of theoretical structure
towards the experiment.
In year 2013-2014 partially supported by PRF grant, we have adopted
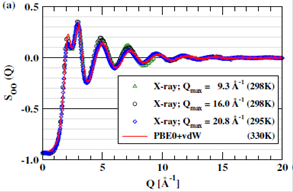
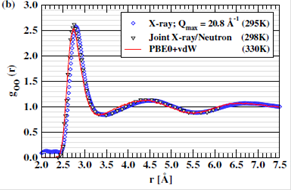
the semi-empirical The results are presented in Fig. 1(a) and (b). From Fig.
1(a), it is clear that the theoretically determined structure factors SOO(Q) is in nearly quantitative agreement with
the experimental results across the entire Q region accessible to the X-ray
scattering experiments, with only a slightly noticeable shift towards higher Q
values. At the same time, the over-structuring in the OO pair correlation
function goo(r) is almost eliminated compared with conventional GGA-AIMD
simulations.