Reports: DNI651680-DNI6: Accurate Ab Initio Studies of Hydrocarbon Physisorption on Carbon Nanotubes
 printer friendly
printer friendly- to obtain similar high-level interaction energies for models representing carbon dioxide adsorption on carbon nanotubes, and to identify the best-performing variants of density functional theory (DFT) and apply them to larger nanotube models,
- to construct a meaningful benchmark set for interactions between carbon dioxide and N-containing polyheterocyclic aromatic hydrocarbons (N-PHACs), models of N-doped nanotubes,
- to obtain an analytical representation of the methane-nanotube interaction potential (fitted to ab initio data) and to apply this potential in simulations of adsorption isotherms and inter- and intramolecular spectra.
Goal 1 has been fulfilled and the corresponding manuscript has just been submitted.The work on Goals 2 and 3 is not completed but significant progress has been made as described below.
1. Models and benchmarks for CO2 adsorption on pristine carbon nanotubes
The gold standard in electronic structure calculations is the coupled-cluster method with single, double, and noniterative triple excitations (CCSD(T)). While this method is quite costly (the largest model for which CCSD(T) can be computed is the coronene-CO2complex), our numerical experience indicates that
(i) no lower-scaling method consistently delivers accuracy comparable to CCSD(T),
(ii) accurate benchmarks can be obtained via the composite MP2/CBS+ΔCCSD(T) approach, utilizing large basis sets at the second-order Møller-Plesset perturbation theory (MP2) level and adding a CCSD(T) correction in a moderate basis,
(iii) to achieve the desired accuracy of 0.1 kcal/mol or better, the ΔCCSD(T) correction has to be computed in at least a partially augmented cc-pVDZ basis.
Accordingly, accurate MP2/CBS+ΔCCSD(T) interaction energies were calculated for 240 complexes involving CO2 and benzene, naphthalene, pyrene, or coronene (flat or curved). Using this benchmark set, a total of 243 combinations of density functionals, dispersion corrections, and basis sets were tested to identify an approximate approach that is not only accurate but performs consistently across different distances, orientations, and model sizes. Unfortunately, the accuracy of all tested approaches deteriorates dramatically in the mildly repulsive region of the interaction (Fig. 1). As DFT interaction energies are consistently too positive in this range, we attributed the problems to an overdamping of the atom-pairwise dispersion expressions (as illustrated in Fig. 2). This finding has led to an exciting new project where we use several model systems (ranging from Ar–Ar to benzene–methane) and accurate asymptotic constants to elucidate the fundamental insufficiency of current damping functions. For the time being, our simple remedy, refitting of the damping parameters, has worked well on the coronene-CO2 benchmark set, leading to several accurate DFT-based approaches that were applied to larger models such as (circum)circumcoronene-CO2 and finite nanotube-CO2 to investigate binding energy convergence with the fragment size.
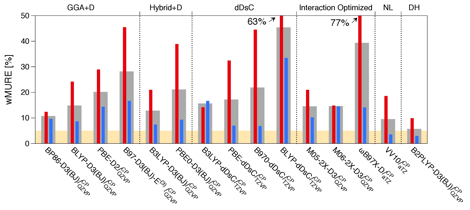
Figure 1. Performance of each density functional, with its best dispersion variant, in the largest basis set. The values presented are mean unsigned relative errors (MURE), weighted to eliminate spuriously large contributions from points close to where the potential crosses zero. Distances shorter than the minimum, longer or equal to the minimum, and all distances are represented by red, blue, and gray bars, respectively.
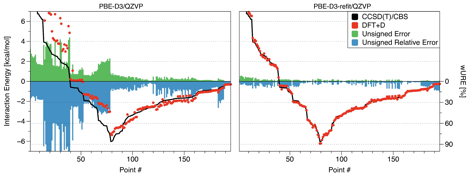
Figure 2. Performance of the PBE-D3/QZVP functional for 195 curved coronene-CO2 geometries, illustrating the overdamping of dispersion at short range alleviated by refitting.
2. Models of N-doped nanotubes and their interactions with CO2
Doping of the carbon nanotubes with nitrogen is an important way of tuning their electronic, optical, and adsorption properties in a broad range. Experimental and simulational studies of N-doped nanotubes indicate two types of local structure: a simple substitution of one carbon atom by a nitrogen and a presence of 3-4 pyridine-like rings surrounding a vacancy. We focus on this latter class of structures and construct a set of model complexes of carbon dioxide with N-containing polyheterocyclic aromatic compounds (N-PHACs) ranging from one to seven rings – see Fig. 3. We consider global-minimum orientations of these complexes (with the CO2 carbon lying in the N-PHAC plane) as well as stacked configurations that may better represent the interaction with an extended nanotube. We again use the MP2/CBS+ΔCCSD(T) approach to compute accurate benchmark interaction energies for a range of systems, orientations, and intermolecular distances, and examine the performance of various DFT variants. An important new aspect is that both DFT and approximate wavefunction-based approaches such as MP2 exhibit a drastically different behavior for the in-plane and stacked structures, confirming that no method below CCSD(T) is consistently reliable. We are currently finalizing the analysis of the DFT performance and plan to employ the best variant in calculations of the interaction energy profile between a CO2molecule and complete 3- and 4-pyridinic vacancies within N-doped nanotubes.
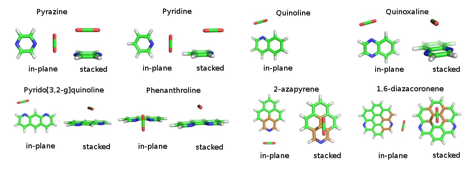
Figure 3. Benchmark complexes of N-PHACs with carbon dioxide.
3. Analytic methane-nanotube potential and its applications
Having established the best DFT variant (BLYP-D3(BJ)/SVP provides an optimal balance between accuracy and performance) and required model size (circumcoronene in case of exterior sites) for accurately recovering methane adsorption energies, we calculated a large number of data points (3705 symmetry-unique configurations per nanotube) and have just fitted the first analytic potential to the exterior data for the (9,0) nanotube. It is sufficient to compute points over a symmetry-unique quadrant of the central circumcoronene ring, as shown in Fig. 4. The analytical potential has a site-site form (with 54 sites on circumcoronene and 5 sites on methane) and combines short-range exponential terms with damped asymptotic contributions. The fit exhibits a mean unsigned error of 0.14 kcal/mol. We are currently testing the quality of the analytical potential and working to combine it with an accurate literature methane-methane potential in grand-canonical Monte Carlo simulations of the methane adsorption isotherm.
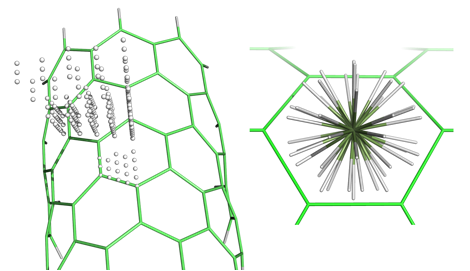
Figure 4. (Left) Symmetry-unique locations of the methane carbon over the nanotube. (Right) A superposition of the angular orientations of methane present in the fitting dataset.










