Reports: DNI352066-DNI3: Cobalt Mediated C-C Bond Transformations toward Small Molecule Synthesis
 printer friendly
printer friendly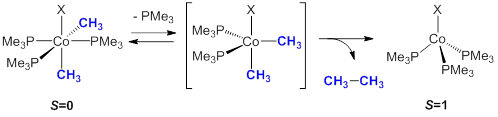
Figure 1.
Under the past year of funding from the Petroleum Research Fund, our program has expanded its attempts to identify the roles that spin state changes and ligand field strengthens have on the rate and selectivity of two-electron C-C bond elimination from six-coordinate cobalt(III) species (Figure 1). In this model for the potential product forming step of C-C cross-coupling, the anionic ancillary ligand X has been targeted for modulation. Two complexes which were targeted for our studies on reductive elimination, X=SPh (1) and X=NCS (2), were serendipitously found to undergo interesting alternative pathways to C-C bond formation, including C-H bond activation of the phenylthiolate and a rare S-atom transfer from the isothiocyanate.
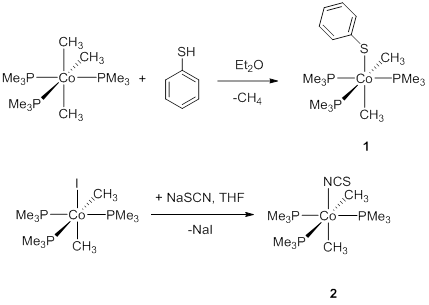
Figure 2.
Complex 1 was prepared by protonation of (PMe3)3Co(CH3)3 in diethyl ether using 1 equiv. of thiophenol, while complex 2 was obtained by alkali salt elimination between (PMe3)3Co(CH3)2and NaSCN (Figure 2). Both complexes were characterized by X-ray diffraction, NMR spectroscopy, and combustion analysis (Figure 3).
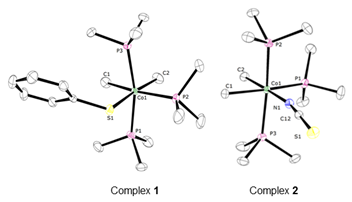
Figure 3.
Thermolysis of the phenylthiolate complex 1 produced only a small amount of the expected ethane product, while affording a more significant amount of methane and the cyclometallated cobalt species, 1b (Figure 4). Each reaction pathway likely initiates from trimethylphosphine dissociation at the position trans to a methyl group to form a five-coordinate intermediate, which has been implicated in a range of other reactions.1
Following C-C bond coupling to generate ethane, two (PMe3)2CoSPh moieties bridge the thiolate ligands to form a dinuclear complex 1a, similar to the observations of Li and co-workers.[2] Interestingly, the presence of a phenyl substituent in proximity to the cobalt in 1 also creates an alternative cyclometallation route for the reaction. Given access to the same five-coordinate intermediate proposed for reductive elimination, the close contact of the ortho C-H bond and the ability to extrude methane from a cobalt-methyl ligand, cyclometallation becomes a highly favorable process. The sequence of reactions that eliminate methane, be it C-H bond oxidative addition to a transient cobalt(V) species or a s-bond metathesis/protonation transition structure, is not discernable with the observations in hand. Notably, when 1-d5 is heated, the degree of cyclometallation was reduced, comprising only 60% of the cobalt products. The identity of the cyclometallation product, 1b was firmly established by solution spectroscopy, and analogy to a previous report from Beck and co-workers who observed this complex from a C-S bond cleavage reaction.[3]
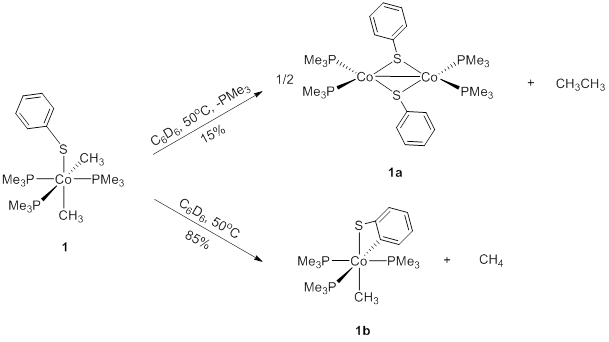
Figure 4.
Complex 2 also deviated from the expected C-C bond elimination reaction, instead exhibiting an unusually S-atom transfer reaction. Upon heating at 50 °C in benzene-d6, a small amount of black precipitate was observed along with formation of ethane and a new doublet located at 1.02 ppm in 1H NMR spectrum and a singlet at 28.4 ppm in 31P NMR spectrum . Analysis of the volatile materials by GC-MS indicated nearly 90% conversion to S=PMe3. The organometallic products proved to be an intractable mixture; however, limited quantities of (PMe3)3Co(CH3)2were observed, consistent with a net S-atom transfer from –NCS (Figure 5). S-atom transfer to trialkylphosphines is a reaction with considerable precedent, but the use of an isothiocyanate fragment as an S-atom source is virtually unknown.[4] The rare ability of this cobalt(III) platform to scission –NCS and promote the oxidation of another substrate could signal a new synthetic pathway for oxidative sulfur additions.
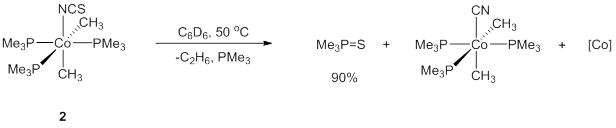
Figure 5.
[1] a) Xu, H.; Bernskoetter, W. H. J. Am. Chem. Soc. 2011, 133, 14956-14959. b) Xu, H.; Williard, P .G.; Bernskoetter, W. H. Organometallics, 2012, 31, 1588-1590. c) Xu, H.; Williard, P .G.; Bernskoetter, W. H. Organometallics, 2013, 32, 798-806.
[2] G. Jiao, X. Li, H. Sun and X. Xu, J. Organomet. Chem., 2007, 692, 4251-4258.
[3] Beck, R.; Frey, M.; Camadanli, S.; Klein, H.F. Dalton Trans., 2008, 4981-4983.
[4] a) Werner, H.; Lotz, S.; Heiser, B. J. Organomet. Chem., 1981, 209, 197-210; b) Werner, H.; Juthani, B. J. Organomet. Chem., 1981, 209, 211-218; c) Donahue, J. P. Chem. Rev., 2006, 106, 4747-4783.










