Reports: ND553496-ND5: Computing Entropies of Adsorbed Molecules
 printer friendly
printer friendlyAdsorbed species on solid surfaces are involved in many reactions of great technological importance, especially in catalysis, electrocatalysis, and fabrication of microelectronics and photovoltaics. As such, there has been tremendous effort worldwide to learn how to predict reaction rates and equilibrium constants for elementary reactions involving adsorbates. Theoretical calculations of rate constants and equilibrium constants for such reactions require knowing both the enthalpy and entropy of the adsorbed species. While much effort has been devoted to measuring and calculating the enthalpies of well-defined adsorbates1, 2, few measurements of the entropies of adsorbates have been reported. We recently reported that all the measured molar entropies of adsorbed molecules are ~2/3 of that for the gas (see Fig. 1), and huge compared to most theoretical predictions currently being used3. Our realization of the reason for this huge entropy led us to propose a new method for theoretically estimating entropies of adsorbed species that should be much more accurate yet still quite easy to implement. These should provide much more reliable predictions of equilibrium constants for surface reactions and the prefactors in their rate constants. 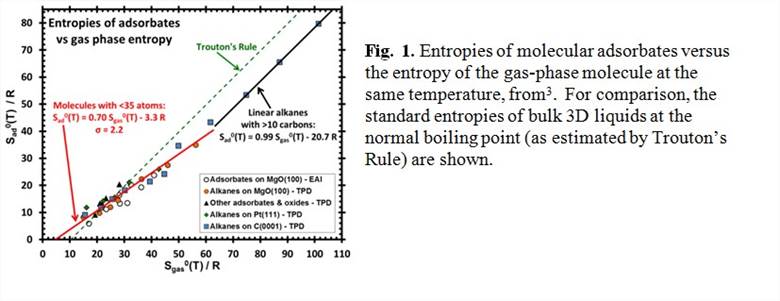
We have made substantial progress in developing this new method. Previously, surface chemists had usually thought about adsorbate entropies in terms of the two limiting cases that have been discussed in statistical thermodynamics texts: the 2D lattice gas model and the 2D ideal gas model 4. In calculating rate constants for surface reactions based on quantum mechanical calculations (mainly density functional theory, DFT) of reactant and transition state energies, surface chemists almost exclusively rely on harmonic transition state theory approaches, which assume that each adsorbate is a localized oscillator with only vibrational modes 5-9. Our recent paper3 proves that these common approximations greatly underestimate the entropies of adsorbed molecules even when they are held together in islands by attractive interactions. We proposed that this is because adsorbates have translations and rotations parallel to the surface that are nearly unhindered. Our new method to accurately consider the nature of the potential energy surface for these motions utilizes an approximation similar to that originally developed by Pitzer and Gwinn10 for calculating partition functions of hindered rotors within gas phase molecules as they transition from harmonic oscillator behavior at low temperatures to free rotor behavior at high temperature. Rotational and translational motions of adsorbates also experience a potential energy that varies sinusoidally with coordinate, and thus the mathematics for solving for their quantum-mechanically allowed energy states and partition functions map directly onto such hindered rotor motion. We use density functional theory with periodic boundary conditions and the nudged elastic band method to estimate rotational and translational barrier heights and their spatial periodicities for prototypical adsorbate/substrate systems. A preliminary result from that method is shown in Fig. 2. 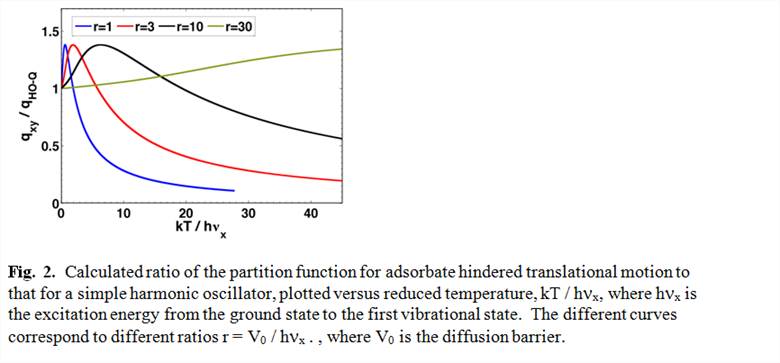
A few model systems with widely varying physical properties are being studied, to learn how differences between systems qualitatively affect entropies and kinetic prefactors for adsorbates.
Educational Benefits
This work is a collaboration with Dr. Líney Árnadóttir, a new Assistant Professor atOregon State University. It provides strong, research-based outreach to a regional university with a less intensive research portfolio than the University of Washington, and guidance for the early-career development of this promising young female investigator. The funding for graduate student support is helping her build up her research group. It partially funded the research of the first three students to join her group: one PhD student, one MS student and one undergrad. This collaboration will allow my students to learn how to perform these types of DFT calculations from her group, and for her group to learn how to do the statistical thermodynamics calculations from my group. This is the first proposal for purely theoretical research I have had, so it adds a much stronger theoretical component to my group, which is known mainly for experimental research.
References:
1. Campbell, C. T.; Lytken, O., Experimental measurements of the energetics of surface reactions. Surface Science 2009, 603, 1365-1372.
2. Brown, W. A.; Kose, R.; King, D. A., Femtomole Adsorption Calorimetry on Single-Crystal Surfaces. Chem. Rev. 1998, 98, 797-831.
3. Campbell, C. T.; Sellers, J. R. V., The Entropies of Adsorbed Molecules. Journal of the American Chemical Society 2012, 134, 18109−18115.
4. Hill, T. L., An Introduction to Statistical Thermodynamics. Addison-Wesley: Reading, MA, 1960.
5. Reuter, K.; Frenkel, D.; Scheffler, M., The steady state of heterogeneous catalysis, studied by first-principles statistical mechanics. Phys. Rev. Lett. 2004, 93, Art. #116105.
6. Gokhale, A. A.; Kandoi, S.; Greeley, J. P.; Mavrikakis, M.; Dumesic, J. A., Molecular-level descriptions of surface chemistry in kinetic models using density functional theory. Chemical Engineering Science 2004, 59, 4679-4691.
7. Honkala, K.; Hellman, A.; Remediakis, I. N.; Logadottir, A.; Carlsson, A.; Dahl, S.; Christensen, C. H.; Norskov, J. K., Ammonia synthesis from first-principles calculations. Science 2005, 307, 555-558.
8. Reuter, K.; Scheffler, M., First-principles kinetic Monte Carlo simulations for heterogeneous catalysis: Application to the CO oxidation at RuO2(110). Physical Review B 2006, 73, 045433.
9. Gokhale, A. A.; Dumesic, J. A.; Mavrikakis, M., On the mechanism of low-temperature water gas shift reaction on copper. Journal of the American Chemical Society 2008, 130, 1402-1414.
10. Pitzer, K. S.; Gwinn, W. D., Energy Levels and Thermodynamic Functions for Molecules with Internal Rotation: 1. Rigid Frame with Attached Tops. J. Chem. Phys. 1942, 10, 428-440.










