Reports: UNI153368-UNI1: Intramolecular Oxidopyrylium Ylide-Silyl Enol Ether [5C+2C] Cycloadditions
 printer friendly
printer friendly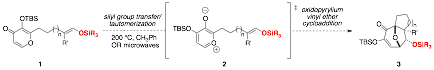
Figure 1. Intramolecular oxidopyrylium ylide-silyl enol ether dipolar cycloaddition.
One of the challenges in using silyl enol ethers as the two-carbon component in the intramolecular oxidopyrylium ylide cycloaddition is the synthesis of silyl enol ethers with defined olefin geometry. If a mixture of (E/Z)-olefin isomers is subjected to the cycloaddition, a diastereomeric mixture of cycloadducts will form (Figure 2).
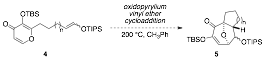
Figure 2. A mixture of (E/Z)-olefin isomers will lead to a diastereomeric mixture of cycloadducts.
Vinyl silanes are an attractive alternative because they are more robust, they can be prepared stereoselectively with defined (E) or (Z)-geometry, and furthermore, the resulting cycloadducts contain alkyl silanes that can be converted to the corresponding hydroxyl group via Tamao-Fleming oxidation.
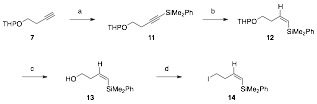
Vinyl silane sidechains were prepared from commercially available but-3-yn-1-ol (1) (Scheme 1). Protection of the alcohol as a THP ether followed by silylcupration afforded the (E)-vinyl silane 8. The THP group was then removed and the resulting alcohol was converted to an iodide to provide (E)-vinyl silane 10.
Scheme 1.a Preparation of (E)-vinyl silane sidechain
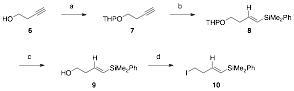
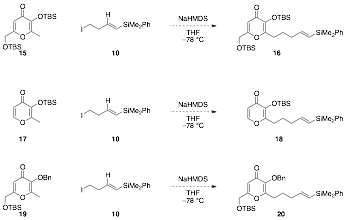
aReagents and conditions: (a) 3,4-Dihydropyran, PTSA, CH2Cl2, rt, 4h, 50%; (b) A similar route was followed for the preparation of the (Z)-vinyl silane 14. Using the same THP ether 7, as described above, the terminal alkyne was silylated to give 11 (Scheme 2). Reduction with DIBAL-H afforded (Z)-vinyl silane 12. Removal of the THP group and conversion of the resulting alcohol to an iodide provided (Z)-vinyl silane 14. Scheme 2.a Preparation of (Z)-vinyl silane sidechain aReagents and conditions: (a) i. nBuLi, THF, –30 °C, ii. Me2PhSiCl, rt, 2h, 84%; (b) DIBAL-H, Et2O, 0 °C, then reflux, 18h, 88%; (c) PPTS, EtOH, rt to 55 °C, 17h, 87%; (d) I2, PPh3, imidazole, CH2Cl2, rt, 6h, 42%. Subsequent efforts to prepare the oxidopyrylium precursor (i.e., 16) via alkylation of various 3-hydroxy-4-pyrone systems with the vinyl silane sidechains were unsuccessful. The alkylation of pyrone derivative 15 (derived from kojic acid) with iodide 10 did not afford any of the desired oxidopyrylium precursor. Similarly, with maltol derivative 17, alkylation with iodide 10 also failed to yield any of the desired product. It is likely that the steric bulk surrounding the nucleophilic site presents a major challenge in the alkylation. The use of a less a sterically hindered substrate (OBn instead of OTBS, 19) was also explored but this was also unsuccessful.