Reports: UR351147-UR3: Control of Selectivity in Intramolecular Carbon-Hydrogen Bond Activations by Transition Metal Complexes
 printer friendly
printer friendly1. Scope and limitation of selective acylation of cycloplatinated complexes
To further understand the regioselective acylation of cyclometalated platinum complexes,1 several cyclometalated platinum complexes were synthesized and their reactions with acetic anhydride and acetyl chloride are shown in Scheme 1.
Scheme 1. Selective acylation of cyclometalated platinum complexes.
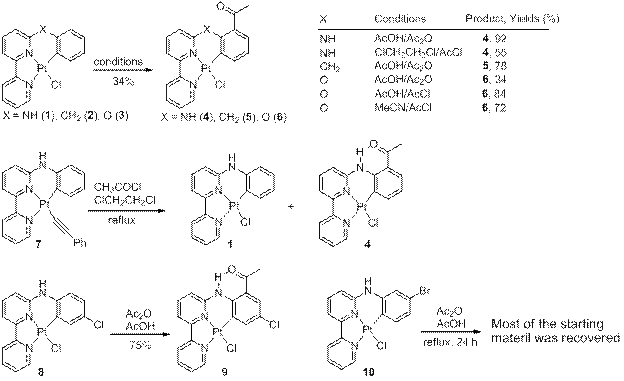
The complexes 2 and 3 were synthesized to evaluate the role of hydrogen bonding in the regioselective acylation reaction.1 The results demonstrated that the acylation did proceed in the absence of hydrogen bonding. Other conditions can also be used for the acylation reaction. For example, the use of acetyl chloride instead of acetic anhydride improved the yield of 6 from 34% to 84% due to the faster reaction and less decomposition. The reaction of phenylacetylide derivative 7 with acetyl chloride in dichloromethane yielded a mixture of 1 and 4 disproved of phenylacetylide ligand. Acylation of complex 8 proceeded smoothly to yield the expected acylation product 9 in good yield, whereas the reaction of 10 under the same conditions did not result in the isolation of an acylated cyclometalated complex. Rather the starting material remained largely unreacted. The underlying factors need to be uncovered. The scope and limitations of this selective acylation are currently under further investigation.
2. DFT studies on the relative stability of isomeric cycloplatinated complexes
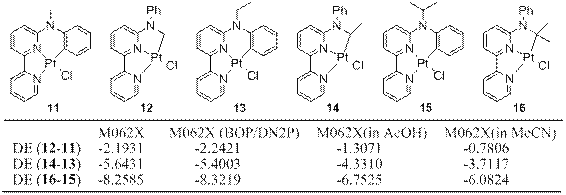
The geometries of platinum complexes 11-16 were optimized with both M062X/def2-TZVP-Pt/cc-pVDZ and BOP/DN2P. The energies of the optimized structures were calculated with M062X/def2-TZVP-Pt/cc-pVDZ and corrected with zero point energy. The results can be summarized as follows: 1) In both gas phase and solvated state, all the sp3 C–H activation products 12, 14 and 16 are predicted to be more stable than their isomeric sp2 C–H activation products 11, 13 and 15, respectively, and the energy difference between the two isomers increases with the increasing size of the substituents from methyl (11 and 12) to ethyl (13 and 14) to isopropyl (15 and 16) (Scheme 2). These results are consistent with the experimental finding2 that the 12, 14 and 16 are the thermodynamic products, and 11, 15 and 17 are kinetically produced; 2) The calculated energies based on the BOP/DN2P optimized geometries are very close to those based on M062X geometries (Scheme 2, columns 2 and 3), indicating that the BOP/DN2P is suitable for a speedy and accurate geometry optimization; 3) Solvents significantly stabilize the cyclometalated complexes (by 11-17 kcal/mol). Acetonitrile stabilizes the complexes by 3-4 kcal/mol more than acetic acid does; and 4) The solvent stabilization on 11, 13 and 15 is greater than that on 12, 14 and 16 by 1-2.5 kcal/mol (Scheme 2, columns 4 and 5).
Scheme 2. Energy difference (ΔE in kcal/mol) between two isomeric sp2/sp3 C-H bond activation products
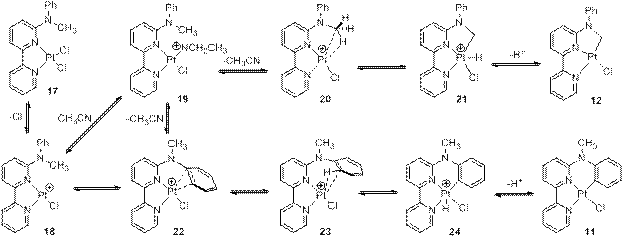
3. DFT studies of reaction mechanisms
DFT calculations were also performed to simulate the reaction pathways for the reaction of N-methyl-N-phenyl-2,2’-bipyridin-6-amine with K2PtCl4 in acetonitrile. The DMol3 program with BOP/DN2P and ;">nudged elastic band (NEB) method were used for the simulations. The initial coordination of the bipyridyl moiety to K2PtCl4 to form the coordination complex 17 was selected as the starting point for the C–H activation. Several of the possible reaction mechanisms for both sp2 and sp3 C–H bond activations are shown in Scheme 3, which involves the agostic interaction as the key intermediate. The π-complex 22 was predicted to be more stable than the agostic complex 23 by 7.0 kcal/mol. The sp2 C-H agostic complex 23 was predicted to be more stable than the sp3 C-H agostic complex 20 by 6.6 kcal/mol.
Scheme 3. Proposed mechanism of C The NEB method was applied to simulate the pathway between 19 and 22 and a minimum energy path was generated with 18 as the intermediate. Calculations predicted tri-coordinated Pt(II) species 18 as a highly reactive species, which either is trapped by the coordinating solvent acetonitrile to form relatively stable complex 19 or collapses to π-complex 22. However, calculations using M062X/def2-TZVP/cc-pVDZ with solvation model PCM (Gaussian 09) indicated that acetonitrile stabilized 18 by 59 kacl/mol, and acetic acid stabilized it by 48kcal/mol. The structures of 17-23 were fully optimized. Further simulations will be performed to understand the controlling role played by the solvent in the selective carbon-hydrogen bond activation. Over the last three years, my research has been mainly supported by the PRF grant. One graduate student and three undergraduates from Dr. Huo’s group have worked on this project. One graduate and one undergraduate from our collaborator Dr. Li’s group were also involved in the research. Four of the students will present their results at the regional ACS meeting SERMACS 2014 in October.
1. Carroll, J.; Gagnier, J. P.; Garner, A. W.; Moots, J. G.; Pike, R. D.; Li, Y.; Huo, S. Organometallics 2013, 32, 4828-4836.
2. Garner, A. W.; Harris, C. F.; Vezzu, D. A. K.; Pike, R. D.; Huo, S. Chem. Commun. 2010, 47, 1902-1904.