Reports: UR652486-UR6: Electron-Induced Reactions: From Methods for Characterization of Intermediates to Mechanisms
Thomas Sommerfeld, PhD, Southeastern Louisiana University
The
research project has two main foci, the development of computational methods
for the characterization of intermediates in electron-induced reactions, and
the characterization of these intermediates themselves. During the second year
(1) work on some specific systems has been finalized, and work on new molecules
has been started, (2) the project on extrapolation methods has been
considerably extended, and (3) a new project regarding the development of
better absorbing potentials has been started.
(1) One
project aimed at studying intermediates was a collaboration with Dr. Robert
Compton's group at the University of Tennessee and Dr. Kit Bowen's group at
John Hopkins University. Negative ion states of large molecules (nitro benzene
moieties coupled to amino acid) were characterized by photoelectron
spectroscopy, collision induced dissociation (by the experimentalists), and
computationally (by us). The combined results were then used to shed light on
the possibility of electron-induced reactions after electron trapping through a
doorway mechanism, and this project is now complete. The second group of
molecules studied were molecules without a dipole but with a large quadrupole
moment. Our results clearly indicate not only that a “critical quadrupole moment” –a notion analogous to the critical dipole moment– does not exist, but also that
electrons bound to this type of molecule are better understood as
correlation-bound than quadrupole-bound species. Hence, our findings have
influenced the anion community's thinking at a fundamental level.
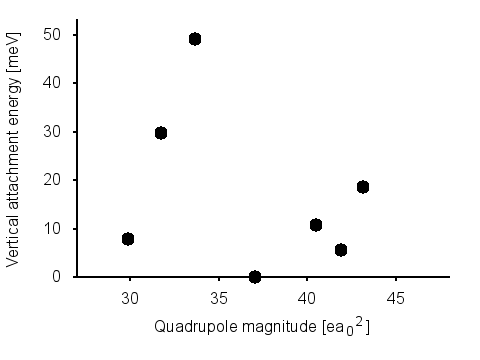
Figure
1: Electron binding energy vs quadrupole magnitude for several molecules with
large quadrupoles but vanishing dipoles. Clearly, there is no trend or cutoff.
(2) The
idea of the extrapolation method is that energy of temporary anions, which are
perhaps from a theoretical viewpoint the most challenging intermediate of
electron-induced reactions, can be obtained from a series of bound state
calculations for a single state. This is done by adding an artificial
stabilizing potential to the Hamiltonian, increasing it until the temporary
state becomes bound, and increasing it further until enough data have been
collected so that one can extrapolate back to zero stabilization.
Unfortunately, any extrapolation as such is to some extent arbitrary, and in
addition, it was widely assumed that extrapolation schemes could only yield
energies, but no lifetimes. As it turns out, in the nuclear physics community
similar methods were independently developed even earlier. These methods do
yield the lifetime, however, they require somewhat different stabilizing
potentials than those typically used in quantum chemistry–a short-range potential as
opposed to long-range Coulomb stabilizations. While a Coulomb stabilization can
be “implemented” by changing the nuclear charges
or adding additional point charges, adding a short-range potential to the
one-electron Hamiltonian in a quantum chemistry code requires implementing
numerical integrals. This has been done, and first applications to a few
well-known resonance states of small molecules look highly promising.
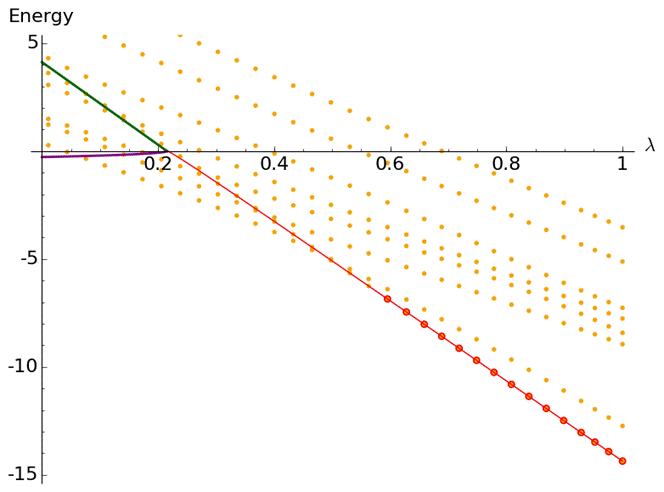
Figure
2: Extrapolation of the energies of the temporary state, once it becomes the
ground state (red), back through threshold, yields the energy (green) and the
decay constant (purple) of the temporary state in the region where it is
unstable.
(3) The
complex absorbing potential method is another way to characterize temporary
anions. Here an imaginary, that is, absorbing, potential is added to the
Hamiltonian, and one follows the eigenvalues of the Hamiltonian into the
complex plane as the strength of the imaginary potential is increased.
Continuum states simply accelerate into the complex plane, whereas temporary
anion states show a pronounced stabilization at their complex energy – the real part is the energy
itself, the imaginary part is the half width, where the lifetime is equal to
the inverse of the width. So far most complex absorbing potential calculations
have used the so called box-CAP, an absorbing potential that has the shape of a
box around the molecule, and then grows quadratically in each Cartesian
direction, because the integrals of this potential can be evaluated
analytically. This functional form itself is adequate for most small molecules
and larger molecules with box-like shapes, but is obviously inadequate in other
cases. More adequate potentials would encase a molecule more closely following
its shape. The disadvantage, however, is again that numerical integrals are
needed. This type of absorbing potential has been implemented, and applied to
several small molecules with p* temporary anions, and these
pilot applications look highly promising.
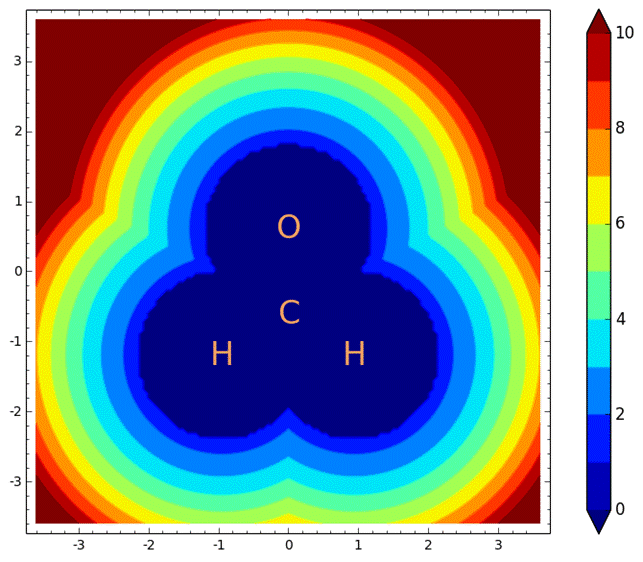
Figure
3: The novel potential used as an absorbing potential.
The work
I can do because of this grant clearly has a huge impact on my career. Without
support for summer research any meaningful professional development at a
primary undergraduate institution would be next to impossible. But especially
in these times of frugality even activities once taken for granted, such as
making presentations at or taking students to regional meetings, become
increasingly difficult. Thus, this grant is key to maintaining an active and
professional research program as such.
For my students essentially
the same is true. It makes a huge difference to know that they do not just
participate in some educational project, but in a real research project that
may at some point land them a poster presentation, a trip to a regional or
national meeting, or even a co-authorship on a paper. They take their work more
seriously, begin to see the value of a deeper level of understanding, and are
more likely to continue “their” research after the mandatory
research classes have been taken.
 printer friendly
printer friendly