Reports: ND653264-ND6: Thermodynamic Insights into Surfactant Assembly in Protic Ionic Liquids
 printer friendly
printer friendlyAt project initiation September 2013 the PI did not have a PhD student enlisted to pursue the proposed research. Over the Fall 2013 semester the PI advertised this project to the incoming first year graduate students in Tulane's Chemical and Biomolecular Engineering program, ultimately recruiting a new student, Kun Chao. Ms. Chao began performing research on this project starting January 2014. Ms. Chao was required, however, to serve as a teaching assistant in her first year to support the classes taught in the PI's department during the Spring 2014 semester. She therefore was not fully supported as a research assistant on this project until the Summer of 2014. Nevertheless, Ms. Chao was able to make substantial progress on this project over the Spring and Summer months, which we describe below.
Beginning in 2014 we began to perform simulations of a series of alkylammonium nitrate (AAN) protic ionic liquids and their mixtures with water to benchmark the thermodynamic properties of existing simulation models for the ionic liquids, as described in the original proposal. Examining the previously reported models for AANs we found a lack of transferability of charges for models with varying alkyl chain lengths, i.e., charges for ethylammonium nitrate (EAN) were not comparable to charges for reported propylammonium nitrate (PAN) or butylammonium nitrate (BAN) models. As a first step then we performed high level ab initio quantum mechanical charge calculations using the GAUSSIAN program to develop a consistent set of transferable charges between alkylammonium nitrates. The charges so derived were then used with the OPLS model for Lennard-Jones, bonded, and torsional interactions to develop a full simulation model. To test the fidelity of this new model at describing the equation of state properties of the AANs we subsequently performed replica exchange molecular dynamics simulations using the program GROMACS to determine the density of these AANs as a function of temperature at atmospheric pressure. The results of these simulations shown in Figure 1 compare quite favorably for EAN, PAN, and BAN giving us confidence in the accuracy of our new model.
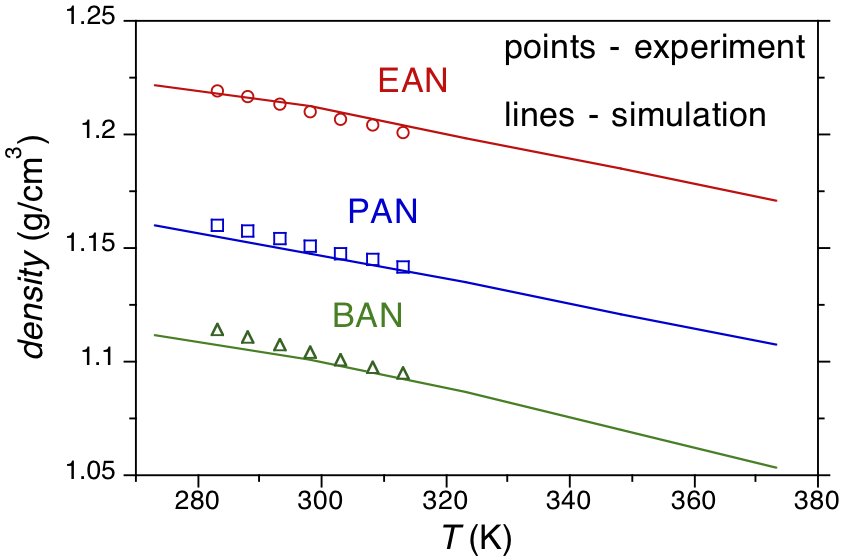
Figure 1. Experimental and simulated densities of EAN, PAN, and BAN as a function of temperature at atmospheric pressure.
Following this test we moved on to examine the ability of the model to describe the volumetric properties of mixtures of EAN with water at 25C. Specifically we performed simulations of EAN with water over a range of concentrations from pure water to pure EAN and determined the mixture density. From the mixture density extracted the excess volumes of mixing and the partial molar volumes of the mixture components (EAN and water) as a function of concentration. The excess volumes extracted from simulation (Figure 2) compare favorably with experiment, showing large negative values over the entire concentration range and a minimum near an EAN mole fraction of 0.3. These observations suggest significant electrostrictive interactions between water and EAN.
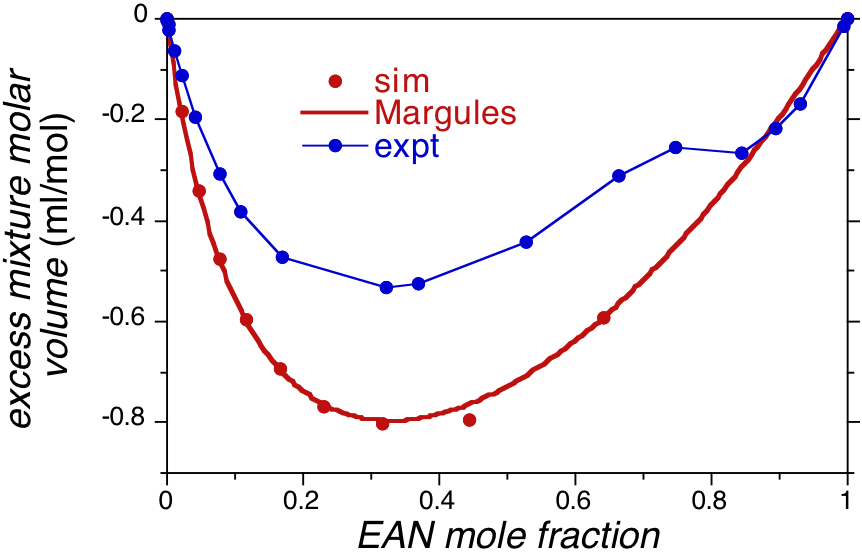
Figure 2. Excess volumes of mixing from simulation and experiment of EAN and water at 25C and atmospheric pressure. Simulation results were fitted to a Margules expansion to extract partial molar volumes.
While the results in Figure 2 are only semi-quantitative, the partial molar volumes extracted from these results show a significantly better comparison between simulation and experiment (Figure 3). Most importantly the results show a nearly linear decrease in the volume of water with increasing EAN concentration, while the EAN volume in roughly constant with increasing water concentration until the concentration hits 75% water. For water fractions above this concentration the EAN volume drops dramatically. This suggests distinct roles for water and EAN in the mixture.
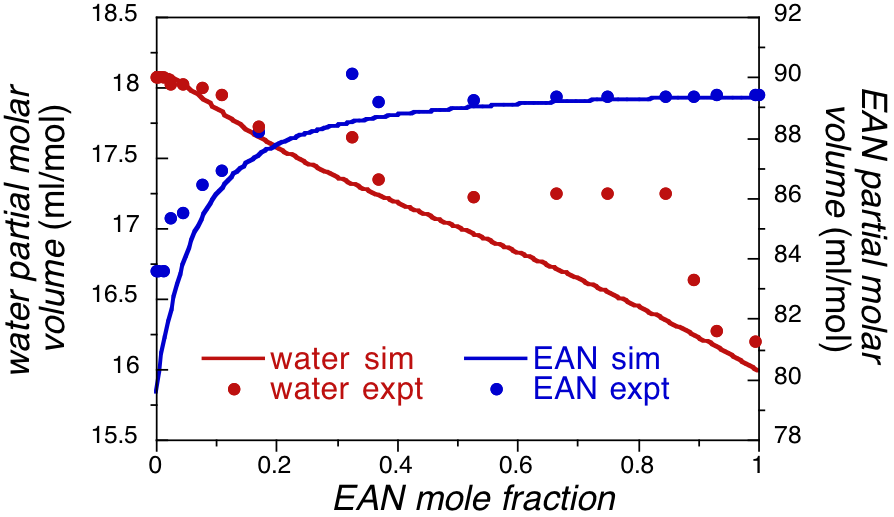
Figure 3. Partial molar volumes of EAN and water mixtures from simulation and experiment.
Further insight can be gained by examining the experimental and predicted x-ray scattering curves for EAN/water mixtures. Specifically, the x-ray plots show two distinct peaks, a primary peak at q values of ~0.5 -1 associated with intermediate range order and a secondary peak at ~1.7 -1 associated with alkyl chain and water interactions. The secondary peak position as a function of EAN concentration is plotted in Figure 4. Overall good agreement is observed between simulation and experiment, indicating the simulations accurately capture the structure of the EAN/water mixture. More importantly the secondary peak position changes most significantly near EAN mole fractions of 0.3, in agreement with the changes seen in the volumetric properties of EAN mixtures reported in Figures 2 and 3. Taken together these results suggest water is swelling the intrinsic structure of the amphiphilic ionic liquid and breaking up aggregates near a concentration of 70% water. We are presently examining these structures in closer detail to get a deeper molecular level understanding in the structural changes observed in EAN/water mixtures. Once these studies have been extended to PAN and BAN mixtures with water we will submit this work for publication.
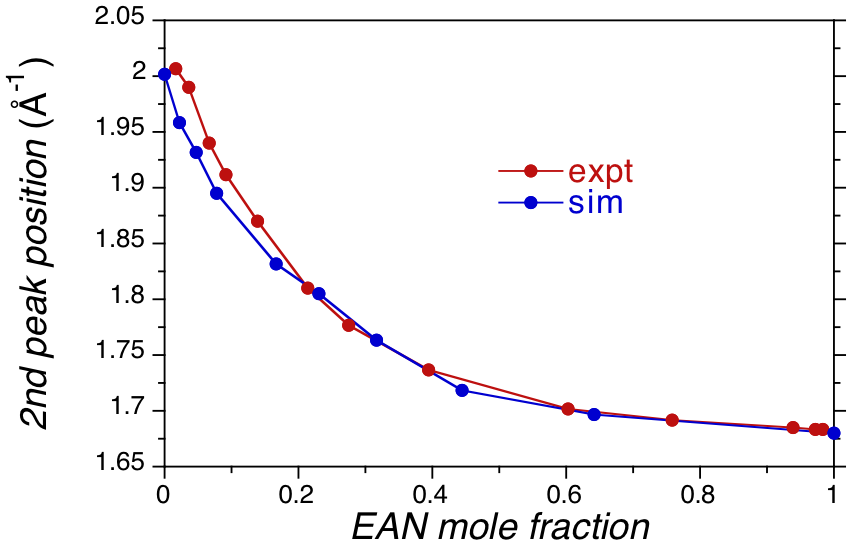
Figure 4. EAN/water mixture secondary x-ray scattering peak position as a function of EAN concentration.
Over the next year we plan to initiate studies on the solubilization of simple non-polar gases in AAN/water mixtures to quantify solvophobic interactions in these mixtures. We will also initiate studies of mixtures of AANs with short chain alcohols as a first step towards examining nonionic surfactant micellization in AANs. This second set of simulations is planned due to a recently published set of systematic scattering experiments of AAN/alcohol mixtures performed by the Warr group from the University of Sydney. This will allow for a more rigorous examination of the forces driving micellar assembly in AANs by simulation.










