Reports: UR653066-UR6: Continued Studies of the Structure, Bonding, and Energetic Properties of Friedel-Crafts Intermediates
 printer friendly
printer friendly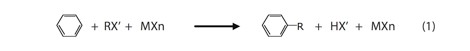
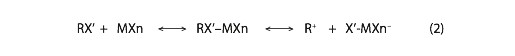
In (1), MXn represents the catalyst, a metal or semi-metal halide that is often AlCl3 or an analogous group III halide (e.g., BF3); though other Lewis acids are also used. RX’ represents the alkyl halide, and the prime on the halogen (X’) distinguishes it from the halogen in the catalyst. We are concerned with the structural and energetic properties of the intermediates in these reactions, alkylhalide-Lewis acid complexes, which are often ionized to some extent, via a process that yields a carbocation and a halogenated Lewis-acid anion (X’-MXn–), e.g.
We are pursuing three primary objectives in our continued work on these systems:
1) Conduct a broad computational survey of RX’–MXncomplexes to determine how changes in M, X, X’, and R affect structural and energetic properties;
2) Determine the energetic favorability of ionization processes involving an additional RX’ molecule, e.g.:
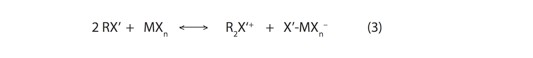
3) Obtain low-temperature IR spectra of RX’/MXn solutions (beginning with RF’/BF3) to provide experimental insight into the ionization pathways (e.g., 2 vs. 3, above)
<p">We have made a great deal of progress on the first of these objectives, and having characterized key trends in a series of RX’–MX3 complexes: M=B,Al; X=F,Cl; R=CH3-,(CH3)2CH-. Initially we made a broad, rather cursory survey of these systems using X3LYP/aug-cc-pVTZ calculations (the preferred method/basis set in our initial work). The most notable observation among these initial results was that the M=Al complexes (e.g., MeCl–AlCl3) were rather strong with binding energies ranging from roughly 10-20 kcal/mol, while by contrast, analogous M=B systems were found to be much weaker with binding energies of less than 5 kcal/mol. The effect of halogen substitution (either X or X’) was more subtle, and in either case changing from F to Cl resulted in a marginal weakening of the complex by a few kcal/mol. The effect of the larger R=(CH3)2CH- group was to increase the binding energies relative to the methylhalide complexes, but the effect was much more notable (3-4 kcal/mol) for the M=Al systems than for the M=B systems (0.5-1.0 kcal/mol). We note also that the overall structural properties parallel the binding energy results. In the weaker complexes, the M-X’ distances are fairly long, and the MX3 subunits are nearly planar. The stronger complexes have shorter M-X’ distances, and MX3units that are significantly distorted from planarity.
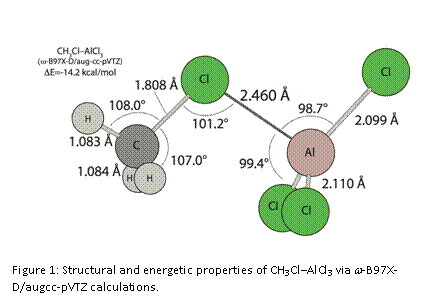
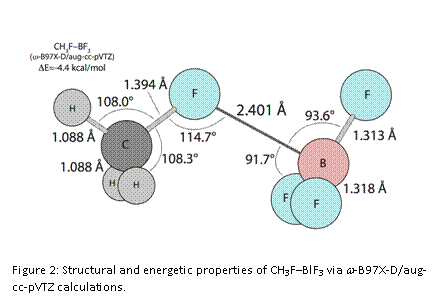
Just recently, we have updated our computational approach by considering a few newer computational methods that are known to provide more reliable energetic results for weaker systems. In the process, we have undertaken a more systematic examination of these RX’–MX3 systems with an eye toward identifying the most stable confirmations. We have established w-B97X-D/aug-cc-pVTZ as our preferred computational approach, as it provides structure and frequency results that are on par with X3LYP/aug-cc-pVTZ, but more reliable energetic results that are key to this project. At this point, we have completed the analyses of the R=CH3-/M=B systems and CH3Cl–AlCl3, and have determined that the eclipsed conformers (as shown in Figures 1 & 2) are the most stable for all these systems. As a whole, the previously identified substituent trends persist with the updated method. We find that CH3Cl–AlCl3 (Figure 1) with a binding energy of 14.2 kcal/mol is a great deal stronger than any of the M=B systems. Furthermore, we note that the AlCl3 subunit is distorted by nearly 10° from its normal, planar configuration, that the C-Cl’ bond is lengthened by about 0.02 Å relative to the value in free CH3Cl, and that NPA charges indicate significant charge transfer from CH3Cl to AlCl3 (about 0.2 e–). The M=B complexes are much weaker, have long, non-bonded B-X’ distances, and exhibit only a few degrees of distortion in the BF3 subunit. CH3F–BF3 is the strongest among these, with a binding energy of 4.4 kcal/mol (Figure 2). With X or X’ = Cl, the result is very weak complexes, with binding energies of 3.6, 2.6, and 2.7 kcal/mol, for CH3Cl–BF3, CH3F–BCl3, and CH3Cl–BCl3, respectively. Thus, the BF3 systems are about 1 kcal/mol stronger than the BCl3systems, but the trend is less clear as X’ is varied.
As a whole, we have made less progress on the experimental goals of the project in the first year, though we have undertaken some key infrastructure improvements, and completed a side project in which we developed some experience with liquid-phase IR techniques. We are currently in the process of rebuilding the optical cryostat system to make improvements that will enable the low-temperature, solution-phase IR experiments, and that apparatus should come on-line by early next year. In the meantime, we identified an opportunity to not only gain valuable experience collecting and analyzing liquid-phase IR spectra (for which optimizing path length and concentration to minimize solvent interference is notoriously difficult), but also engaged an entire cohort of students in our capstone physical/analytical lab in a research-based project. This involved an IR study of alcohol/solvent interactions, and we ultimately observed the OH stretching bands (fundamentals and first two overtones) of five alcohols in ten different solvents with varying degrees of interaction with the hydrophilic OH group of the alcohols. The frequency shifts provide insight into the relative strength of the alcohol-solvent interactions, and the overtone data enable guage variations in anharmonicity that accompany weak H-bonding interactions. In the next year, we will submit these results for publication, and students from the course will most likely be co-authors on that manuscript. Furthermore, we gained insight into key design parameters needed to deign the cell for IR studies of RX/MXn mixtures.










