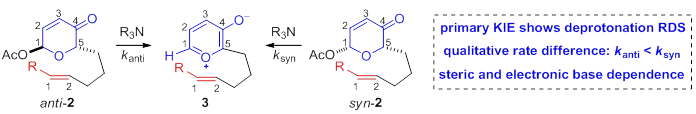
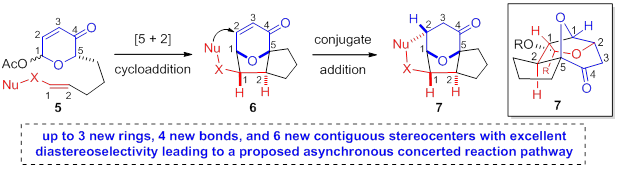
Reports: UNI152391-UNI1: Stereoselective N-Heterocyclic Carbene (NHC) Catalyzed Oxidopyrylium-Enolate [5+2] Cycloadditions towards Bridged, Polycyclic Ethers
 printer friendly
printer friendly
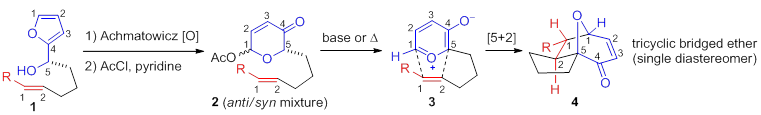
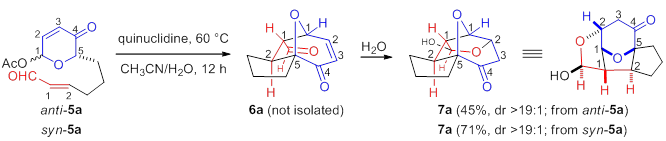
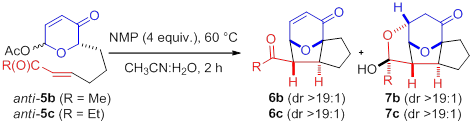
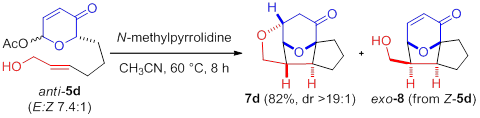
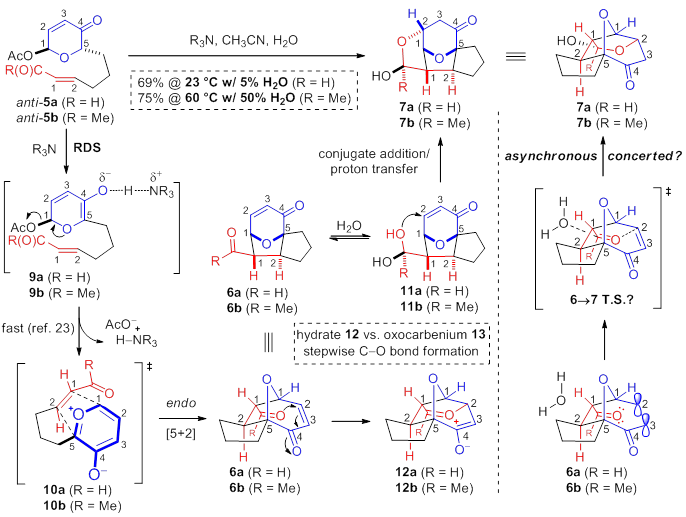
Compared to the facile conversion of the aldehydes, conjugate addition of pyranone-ketones 6b,c derived from acetoxypyranone-enones 5b,c was non-spontaneous. Interestingly, upon addition of water (5%), trace lactol 7b was observed (entry 1). Increasing the concentration of water proved to be sufficient to afford the desired lactol 7b (entry 2). Similarly, acetoxypyranone-enone 5c afforded the analogous lactol 7c albeit with slightly decreased conversion (entry 3).
In an effort to extend
the scope, we treated acetoxypyranone-enal 2a with a variety of alcohols
to afford caged acetal-ethers 7e-l. Utilization of excess methanol (CH3CN:MeOH
95:5) with 3Å molecular sieves to ensure the exclusion of water provided acetal
7e as a single diastereomer in 63% yield (entry 1). Additional
functionalized primary alcohols afforded similar results (entries 2-4). More
hindered alcohols gave mixed results; isopropanol delivered acetal 7i in
moderate yield (entry 5), but tert-amyl alcohol afforded a complex
mixture with no acetal 7j detected (entry 6). Although p-cresol
gave poor yield, acetal 7k represents an interesting result arising from
a different hydroxyl functionality (i.e. phenol) attacking the presumed
pyranone-aldehyde 6a (entry 7). Finally, Boc-L-serine methyl ester
provided acetal 7l in good yield as an expected mixture of inseparable
diastereomers (entry 8). Entry ROH time (h) 7e-l (% yield) 1 MeOH 6 63 (e) 2 H2C=CH(CH2)4OH 6 53 (f) 3 p-MeO(C6H4)CH2OH 1 62 (g) 4 HC≡CCH2OH 2.5 57 (h) 5 i-PrOH 4 53 (i) 6 2-methyl-2-butanol 6 ND (j) 7 p-Me(C6H4)OH 2.5 20 (k) 8 Boc-L-serine methyl ester 1 73 (l)