Reports: UNI551724-UNI5: Surface Immobilization of Cobalt-Diglyoxime Complexes: The Effects of Interfacial Chemistry on Electrocatalytic Activity in Aqueous Media
 printer friendly
printer friendlyFigure 1 depicts the immobilization strategy that was originally envisioned for the cobaloxime catalysts. In the first annual reporting period, surface electrochemistry, UV-vis spectroscopy, elemental analysis, and steady-state fluorescence demonstrated that the axial pyridine ligand is highly labile under the solvent conditions desired for catalysis. For this reason, ligation to surface pyridine groups was abandoned as an immobilization strategy. Instead, non-specific hydrophobic interactions were used to achieve cobaloxime immobilization. During the present reporting period, this same immobilization strategy has been used to test the catalytic activity of cobaloximes. In brief, hydrophobic films are prepared by the electrochemical reduction of 4-hexyl-benzenediazonium. These modified surfaces are incubated in an organic solution of cobaloximeleading to retention of the compound within the surface film. After this modification, the electrodes are suitable for electrochemical hydrogen production at low pH.
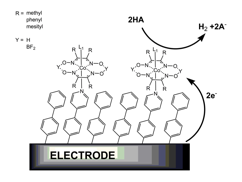
Figure 1. A schematic representation of the immobilization strategy originally pursued. The axial pyridine ligand proved too labile for satisfactory hydrogen production.
Catalytic hydrogen production proceeds following protonation of a low-valent state of the metal. In hopes of extending the catalytic activity to higher pH, we are introducing electron-donating groups on the glyoxime ligand. The premise is that, for a given oxidation state, the electron-donating substituent will make the cobalt ion more basic, favoring protonation and catalytic turnover under less acidic conditions. For this reason, the laboratory largely has transitioned to a study of cobalt amidoxime complexes. The first ligand examined in this series was bis(N-hexylamidoxime). The BF2-capped cobalt complex prepared from this ligand, Co(dNhgBF2)2, is shown in Figure 2. The metallation reaction proceeds in a manner similar to "typical" methyl- or phenyl- glyoximes. However, a higher temperature is required to achieve astable complex. Refluxing hexanes were used in place of room temperature diethyl ether.
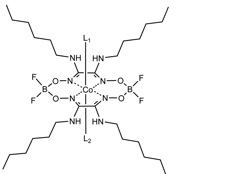
Figure 2. Structure of Co(dNhgBF2)2, a cobalt amidoxime derivative.
The as isolated Co(dNhgBF2)2 was found to be a Co(III) complex, rather than Co(II) as is typical for BF2-capped cobaloximes. It is presumed that the increased electron density on the cobalt ion makes the Co(II) state sufficiently reducing to react with molecular oxygen during the aerobic synthesis. Consequently, the Co(III) complex is isolated. In comparison to the metal-free ligand, Co(dNhgBF2)2 showed characteristic shifts in the C=N and N-O vibrational frequencies following complex formation. The diamagnetic Co(dNhgBF2)2 allowed for analysis by 1H- and 13C- NMR spectra, with surprisingly few spectral changes upon metallation other than loss of the signal for the oxime protons. Figure 3 compares the UV-vis spectra of Co(dNhgBF2)2, to spectra for common cobaloximes. There is a bathochromic shift in the MLCT band of the amidoxime derivative. Figure 4 shows cyclic voltammograms of Co(dNhgBF2)2 recorded in acetonitrile solution. The Co3+/2+ and Co2+/1+ couples show pronounced shifts to cathodic potentials in comparison to a typical cobaloxime. Taken together, these results are consistent with a substantial electron donating effect of the amidoximeligand. As desired, this modification results in more negative reduction potentials for metal-based redox processes.
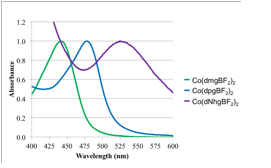
Figure 3. UV-vis spectrum of the cobalt amidoxime complex Co(dNhgBF2)2, compared to spectra of the well-known methyl and phenyl cobaloxime.
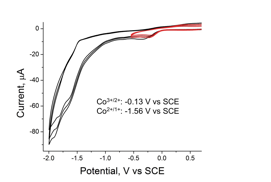
Figure 4. Cyclic voltammograms of Co(dNhgBF2)2 recorded in an acetonitrile solution.
Work with cobalt amidoximes was undertaken in order to shift the cobalt-based redox processes to more negative reduction potentials. The results from Co(dNhgBF2)2 suggest this is possible. However, in the present case, the cathodic shift is so extreme that the Co3+/2+ couple operates at nearly the same potential as the Co2+/1+ couple of the well-known phenyl cobaloxime. Electrochemical analysis of surface immobilized catalysts in water found little difference in their pH optima, with both compounds operating only under acidic conditions. Attempts to access hydrogen production at high pH using the Co(I) state of Co(dNhgBF2)2were complicated by substantial background currents at these very negative potentials.
In light of the results with Co(dNhgBF2)2, we have undertaken a more systematic study of cobalt amidoxime catalysts. To investigate the effect of alkyl chain length, a series of methyl,N,N-dimethyl, ethyl, N-ethyl, hexyl, and N-hexyl glyoxime ligands is being prepared. Our results with the corresponding cobalt complexes suggest that ethyl and hexyl derivatives behave similarly to traditional dimethylglyoxime. We are still working with N,N-dimethyl and N-ethyl derivatives to expand our knowledge of cobalt amidoximes beyond Co(dNhgBF2)2. We also are preparing a series of N-aryl glyoximes in hopes of moderating the electron donating ability of the amidoxime ligand. By tuning substituents on the N-aryl ring, it seems feasible to tune the Co2+/1+couple to a modestly negative potential suitable for hydrogen production at high pH. Due to an extension of the funding period, work along these lines will be ongoing.
During the current reporting period, PRF funding has provided for materials and supplies, as well as enabling six undergraduate researchers to participate in this work through funded positions. This support has had a substantial, positive impact on the output of my laboratory and the education of my research students. Data from this project has been used in multiple grant proposals and conference presentations. Over the course of this grant, four undergraduates working on the project have graduated, and three of those have gone on to PhD programs. The availability of PRF funding has allowed undergraduates to devote greater time to research, while increasing their own valuation of their research efforts. This has increased the productivity of my laboratory.










