Reports: ND552696-ND5: Two-Dimensional Nanoporous Self-Assembled Molecular Layers: New Structures for Selective Adsorption and Catalysis
 printer friendly
printer friendly
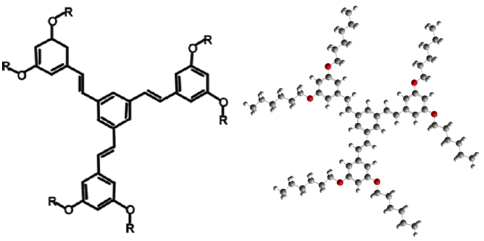
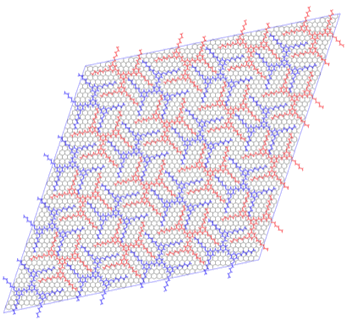
Spontaneous molecular self-assembly is a promising route for bottom-up manufacturing of two-dimensional (2D) nanostructures with specific topologies on atomically flat surfaces. Of particular interest is the possibility of selective lock-and-key interaction of guest molecules inside cavities formed by complex self-assembled two-dimensional host structures. Understanding of these systems could lead to the design of materials with highly specific selective adsorption, or catalysis of chemical compounds. Our studies focus on structures constructed with molecules of 1,3,5-tristyrylbenzene Recent real-space imaging of reorientation of molecules adsorbed inside nanocavities formed in submonolayer films of such molecules show their potential as building blocks for self–assembled nanostructures [1-6]. Our approach focuses on understanding the mechanics of these structures from first principles. A triangular super-lattice of TSB3,5-C6 molecules (4 x 4 cluster, ~ 12.5 nm sides, commensurate, at an angle of 11.7¡ to the REFERENCES
8. M. Head-Gordon, J.A Pople, and M.J Frisch, Chem. Phys. Lett