Reports: DNI151707-DNI1: Novel Catalytic Enantioselective [3,3]-Sigmatropic Rearrangements for the Preparation of Highly Substituted Fused Heterocycles and Biaryls
Laszlo Kurti, PhD, University of Texas Southwestern Medical Center at Dallas
The
growth of knowledge in the chemical sciences has seen a dramatic increase since
the beginning of the 21st century. The advance is particularly
significant in synthetic chemistry because of its centrality and value to
society. Key to all of this is the constant development of novel C-C and
C-heteroatom bond-forming strategies, methods and reagents that expand the
toolbox of synthetic organic chemists and enable the environmentally friendly
construction of complex molecular structures using the fewest number of
chemical steps and generating the least amount waste. At the heart of our
research program is the discovery, mechanistic understanding and exploitation
of new patterns of reactivity. We aim to design/create synthetic strategies
that previously did not exist and enable the practitioner to prepare both
existing as well as new structurally complex building blocks/scaffolds quickly
and more efficiently than before. The Kürti group is pursuing the development of powerful and
practical new methods for the synthesis of N- and O-heterocycles.
Ideally these new routes are expected to: (a) be operationally simple; (b)
utilize readily available and inexpensive starting materials; (c) avoid the use
of TM catalysts, ligands and strong oxidizing agents; (d) achieve C-X
bond-formation with controllable and high regioselectivity; (e) build molecular
complexity rapidly and with exceptional step-economy (i.e., in one-pot); (f)
permit the presence of functional groups that are generally not compatible with
TMs (i.e., halogens, that allow further functionalization via the
cross-coupling manifold) and (g) be complementary to existing cross-coupling
methods by allowing the facile preparation of currently unknown or otherwise
inaccessible heterocycles. The
carbazole ring system appears as a motif in a large number of biologically
active natural products, active pharmaceutical ingredients (APIs) and novel
functional organic materials. Given the tremendous importance of functionalized
carbazoles and their derivatives across many fields, it is not surprising that
during the past 50 years dozens of synthetic methods have been developed to
access this valuable heterocyclic framework with a variety of substitution
patterns. However, the overwhelming majority of these methods require harsh
reaction conditions which are incompatible with many sensitive functional
groups. Thus, we have developed a
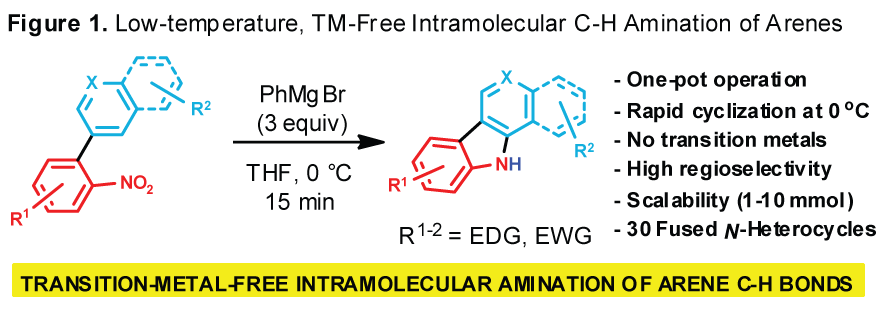
low-temperature, transition metal-free, rapid and highly regioselective
intramolecular amination of arene C(sp2)-H bonds, starting from
readily available 2-nitrobiaryls (Figure 1). This transformation has a wide scope as demonstrated by the preparation of a
total of 30 fused N-heterocycles, including two bioactive carbazole
alkaloids. A preliminary examination of the mechanism using DFT suggests that a stepwise electrophilic aromatic
cyclization mechanism may be operative. We anticipate
that this transformation may serve as a prototype for related powerful
transformations that build molecular complexity rapidly, under mild conditions
with exceptional step-economy and in an environmentally friendly fashion.
The
benzofuran structural motif, in particular benzo[b]furan, appears widely
in many biologically active natural products, active pharmaceutical ingredients
(APIs), and intermediates/ building blocks for the synthesis of complex
molecules as well as in functional materials such as dyes, polymers and
film-forming compounds.
Given
the importance of functionalized benzo[b]furans and their derivatives
across several key scientific fields, there is great demand for versatile
synthetic methods that allow their rapid, efficient and readily scalable
preparation. Thus, it is not surprising that during the past few decades quite
a few synthetic approaches have been developed to access these heterocycles
from acyclic precursors. The overwhelming majority of these methods utilize
transition metal (TM) catalysts (e.g., Cu, Pd, Pt) to form the required C-C and
C-O bonds of the benzo[b]furan substructure; most commonly the furan
nucleus is assembled on a pre-functionalized benzenoid scaffold (i.e., via
cycloisomerization). Even though TM-catalyzed syntheses of benzofurans are
currently the most widely used, they often suffer from certain drawbacks
such as necessity to employ harsh conditions (e.g., elevated temperatures for
extended periods of time) and generation of a toxic heavy metal waste that is
expensive to remove especially during the production for APIs in which residual
metal contamination needs to meet stringent specifications.In
light of the above, there is a clear need for the development of versatile
TM-free approaches to functionalized benzo[b]furans that utilize
structurally diverse, readily available/inexpensive starting materials and/or
reagents.
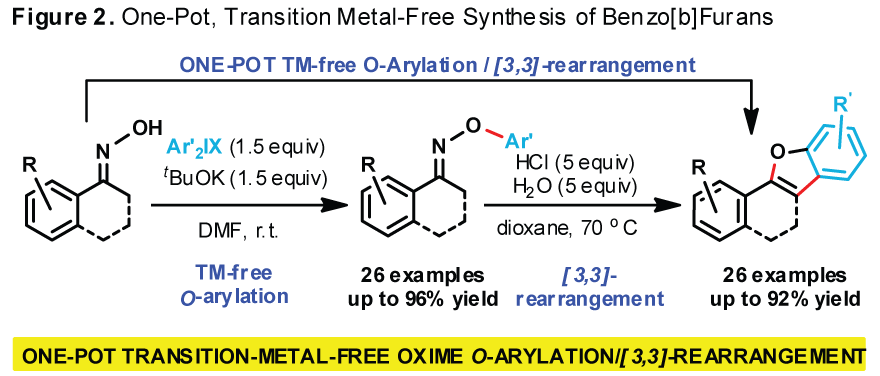
We
have developed a scalable, transition-metal-free, direct O-arylation of
ketone oximes utilizing structurally diverse and readily available
diaryliodonium salts as electrophilic arylating agents (Figure 2).
The O-arylation proceeds under mild conditions at ambient temperature
and furnishes the corresponding O-aryl oximes in high yield while
tolerating both electron-rich and electron-poor substituents on both reactants.
The O-arylated ketoxime products can be efficiently converted into
substituted benzo[b]furans either under acid-mediated or N-acylation
condition if acid-sensitive functionalities are present. We have also developed a one-pot
sequence that conveniently affords benzo[b]furans in good isolated
yields, while avoiding the preparation and handling of sensitive O-aryloxyamines.
Aziridines,
the three-membered and equally highly-strained nitrogen analogues of epoxides,
are important synthetic intermediates (i.e., building blocks) en route to
structurally complex molecules due to their versatility in myriad regio- and stereoselective
transformations (ring-openings and -expansions as well as rearrangements). The
aziridine structural motif, predominantly N-H and to a lesser extent N-alkyl,
also appears in natural products which exhibit potent antibiotic,
immunomodulatory and anticancer properties. Current direct olefin aziridination
methods rely either on the transfer of substituted nitrenes, generated using
strong external oxidants, to the C=C bond of olefins or the transfer of
substituted carbenes to the C=N bond of imines. Normally, the result is an
aziridine bearing a strongly electron-withdrawing N-protecting group;
removal of these protecting groups is problematic as it often results in the
undesired opening of the aziridine. In addition, the high reactivity of these N-protected
nitrenes can give rise to non-productive allylic C-H amination as well as the
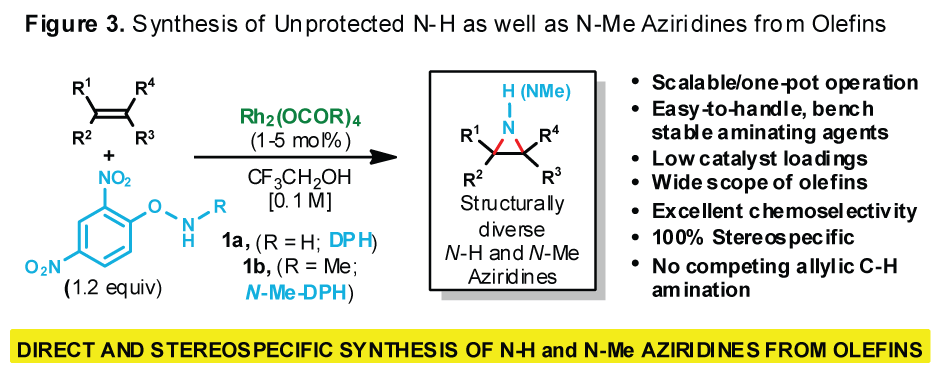
loss of stereospecificity.We have developed a mild,
versatile method for the direct stereospecific conversion of structurally
diverse mono-, di-, tri-, and tetrasubstituted olefins to N-H aziridines using
O-(2,4-dinitrophenyl)hydroxylamine (DPH) via homogeneous rhodium catalysis with
no external oxidants. This method is operationally simple (i.e., one-pot),
scalable, and fast at ambient temperature, furnishing N-H aziridines in
good-to-excellent yields. Likewise, N-alkyl aziridines are prepared from
N-alkylated DPH derivatives. Quantum-mechanical calculations suggest a
plausible Rh-nitrene pathway.
 printer friendly
printer friendly